Psoriatic arthritis (PsA) is a typical complication of psoriasis (PSO) that is often accompanied by nail PSO, vertebral and/or pelvic involvement, enthesitis and iritis. PsA leads to the destruction and/or ankylosis of the peripheral joints or spine, resulting in poor quality of life.1 Interleukin (IL)-23, IL-17 and tumour necrosis factor (TNF) play a pivotal role in the pathogenesis of PsA and are targets for treatment.2 Biological disease-modifying antirheumatic drugs (bDMARDs), which target these cytokines, and targeted synthetic DMARDs (tsDMARDs), which target Janus kinases, have become available and are widely used in clinical practice.3–19 Despite the availability of bDMARDs and tsDMARDs, which have different mechanisms of action with different targets, there are still unmet needs: for example, disease activities are often less responsive to these existing bDMARD and tsDMARD treatments and relapse and/or develop drug intolerance. The emergence of new drugs is expected to increase the number of effective treatment options for these patients. Recently, data on the efficacy and tolerability of bimekizumab, a humanized monoclonal immunoglobulin (Ig) G1 antibody against IL-17A and IL-17F, have been reported.20–24 This article reviews the results of the two latest phase III clinical trials of bimekizumab (BE OPTIMAL and BE COMPLETE) and its promising role in clinical practice for the treatment of PsA.23,24
Bimekizumab, a selective inhibitor of interleukin-17A and interleukin-17F
IL-17A is the most famous member in the IL-17 family, and its structure is 50.0% homologous to IL-17F. After the homo- or heterodimers of IL-17A and IL-17F are secreted, both signals are transmitted via the IL-17RA and IL-17RC receptor complexes.25 Various immune cells involved in the pathogenesis of PsA secrete IL-17A and IL-17F. However, IL-17A is mainly produced by Th17 cells, and IL-17F is mainly produced by other immune cells, such as CD8+ T cells, natural killer T cells, lymphoid tissue inducer-like cells, innate lymphoid cells and γδT cells.25 Despite their high homological structure, IL-17A and IL-17F exert distinct pro-inflammatory effects, and each has a synergistic effect with TNF to amplify the inflammatory response. In fact, translational research in rheumatoid arthritis has demonstrated that IL-17F synergistically induces a pro-inflammatory gene signature with TNF, similar to the combination of IL-17A with TNF.26,27 Therefore, it has been suggested that both IL-17A and IL-17F can be targeted to control PsA.26,27
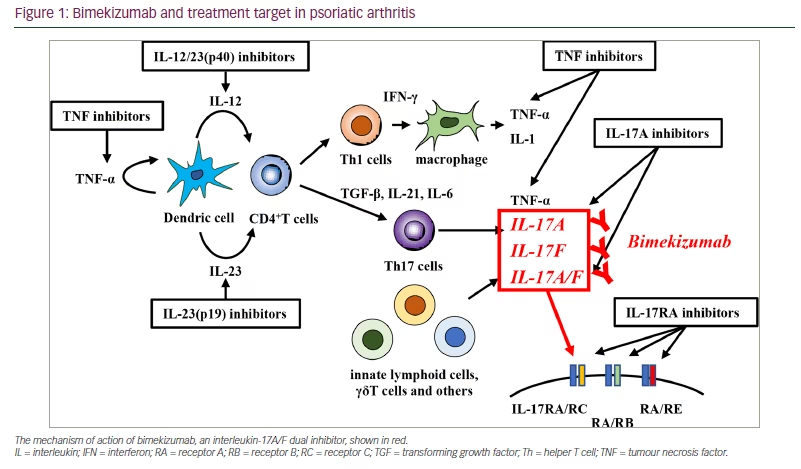
Bimekizumab is a humanized monoclonal IgG1 dual antibody that selectively and directly inhibits both homo- and heterodimers of IL-17A and IL-17F by binding to structurally homological sites on IL-17A and IL-17F and dually inhibiting (Figure 1).28 In contrast, brodalmab is an anti-IL17 receptor A monoclonal antibody that inhibits the activities of IL-17A, IL-17F, IL-17A/F, IL-17C and IL-17E.29 Furthermore, the dual inhibition of IL-17A and IL-17F by bimekizumab suppresses pro-inflammatory gene expression and cytokine production in vitro more efficiently than the specific inhibition of IL-17A or IL-17F individually.30 Additionally, a preclinical study on the osteogenic differentiation of human periosteum-derived cells revealed that the dual inhibition of IL-17A and IL-17F may modulate osteoblast activity to a greater extent than IL-17A inhibition alone.30
Bimekizumab in moderate-to-severe plaque psoriasis
In phase III and IIIb studies, bimekizumab showed tolerability and a statistically significant superior Psoriasis Area and Severity Index (PASI) 90 response rate and Investigator’s Global Assessment 0/1 achievement compared with the placebo (BE READY; ClinicalTrials.gov identifier: NCT03410992),31 ustekinumab (BE VIVID; ClinicalTrials.gov identifier: NCT03370133)32 and adalimumab (BE SURE; ClinicalTrials.gov identifier: NCT03412747)33 in patients with moderate-to-severe plaque PSO. It also showed a statistically significant superior PASI 100 response rate compared with secukinumab (BE RADIANT; ClinicalTrials.gov identifier: NCT03536884), observed as complete skin clearance.34
Methods
In our literature search, PubMed was used to collect studies assessing the efficacy of bimekizumab in patients with PsA. The search was limited to include all studies indexed in PubMed between January 2020 and January 2023. The keywords used in the search were “PsA”, “psoriatic arthritis“, “bimekizumab” and “treatment”. Moreover, the search was restricted to randomized controlled/clinical trials and studies published in English.
The identified studies were subsequently entered into an abstract screening sheet. The study was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines.
All studies identified in the original literature search were evaluated for eligibility using the Patients, Intervention, Comparison and Outcome approach:
- Patients: sex was not specified; age was not specified; disease/condition was specified as PsA and spondyloarthritis; geographic requirements were not specified; others were not specified
- Interventions: bimekizumab
- Comparison/control: placebo
- Outcome for PsA: achieving at least 20%, 50% and 70% in the American College of Rheumatology (ACR) response criteria (ACR20, ACR50 and ACR70, respectively); Disease Activity Index for Psoriatic Arthritis; PASI; Minimal Disease Activity; Simplified Disease Activity Index; Health Assessment Questionnaire (HAQ); Medical Outcomes Study Short-Form 36-Item Health Survey (SF -36); van der Heijde–Sharp score.
Results
As a result of our review, four full-text articles were included in the initial abstract screening. All four articles were included in the full-text screening and subsequently used to conduct the narrative review.21–24
Bimekizumab in active psoriatic arthritis: BE ACTIVE, a dose-ranging phase IIb clinical trial
BE ACTIVE was a 48-week randomized, double-blind, placebo-controlled, dose-ranging phase IIb study (ClinicalTrials.gov identifier: NCT02969525), which included patients aged ≥18 years with active PsA (≥6 months disease duration, tender joint counts [TJC]/swollen joint counts [SJC] ≥3).20 It revealed the safety profile and tolerability of bimekizumab and that the primary endpoint of achieving at least 50% in the American College of Rheumatology (ACR) response criteria (ACR50) response rate was higher at Week 12 compared with placebo.20 A total of 206 patients were randomly assigned (1:1:1:1:1) to receive the placebo, 16 mg bimekizumab, 160 mg bimekizumab, 160 mg bimekizumab with a one-off 320 mg loading dose or 320 mg bimekizumab every 4 weeks. After 12 weeks, the patients assigned to the placebo and 16 mg bimekizumab groups were randomly reassigned (1:1) to either the 160 mg or 320 mg bimekizumab group. The primary endpoint was the proportion of patients achieving an ACR50 response at week 12. At week 12, achieving an ACR50 response was significantly higher in the 16 mg bimekizumab group (odds ratio [OR] 4.2, 95% confidence interval [CI] 1.1–15.2; p=0.032), 160 mg bimekizumab group (OR 8.1, 95% CI 2.3–28.7; p=0.0012) and 160 mg bimekizumab group with a loading dose (OR 9.7, 95% CI 2.7–34.3; p=0.0004) compared with the placebo group. Moreover, at week 12, the incidence of treatment-emergent adverse events (TEAEs), most of which were mild to moderate, was 24/42 (57%) in the placebo group and 68/164 (41%) in the bimekizumab group. Serious TEAEs were identified in nine patients, eight of whom received bimekizumab. No deaths or reports of inflammatory bowel disease were identified.
The efficacy of long-term bimekizumab treatment on patient-reported outcomes was also demonstrated in the BE ACTIVE-104-week open-label extension study (160 mg bimekizumab every 4 weeks).21 This study assessed the arthritis pain visual analogue scale, PsA Impact of Disease-9, SF-36 and HAQ-Disability Index (HAQ-DI) scores. They found that improvements in pain and fatigue were sustained from weeks 48 to 152.
Bimekizumab in active psoriatic arthritis, naïve to biologics: BE OPTIMAL, a phase III clinical trial
BE OPTIMAL is a 52-week, multicentre, randomized, double-blind, placebo-controlled, active reference (adalimumab) phase III trial (ClinicalTrials.gov identifier: NCT03895203).23 Patients aged ≥18 years with adult-onset active PsA (≥6 months disease duration, TJC/SJC ≥3) and one or more active psoriatic lesions and/or a documented history of PSO were eligible for this trial. Patients with a current or previous history of bDMARDs treatment for PsA or PSO were excluded. Patients who completed week 52 and met the inclusion criteria were enrolled in the open-label extension study that is currently in progress (BE VITAL; ClinicalTrials.gov identifier: NCT04009499) at week 48, after which they received 160 mg bimekizumab every 4 weeks.35 In total, 852 patients were randomized 3:2:1 as follows: 431 patients were administered subcutaneous bimekizumab 160 mg every 4 weeks, 281 were administered a placebo every 2 weeks, and 140 were assigned to the reference group (40 mg adalimumab every 2 weeks).
The primary endpoint was the proportion of patients achieving an ACR50 response at week 16 (bimekizumab compared with placebo).23 This study was not designed to compare adalimumab with other groups (bimekizumab or placebo); however, an adalimumab reference group was used to assess the benefit–risk profile of bimekizumab and mask active treatment. Discontinuation rates were low (29/852 [3.4%]), and 821/852 (96.0%) patients completed week 16 of the assigned treatment (bimekizumab 414/431 [96.0%]; placebo 271/281 [96.4%]; adalimumab: 136/140 [97.1%]), whilst 806/852 (95.0%) completed week 24 (bimekizumab 404/431 [93.7%]; placebo 269/281 [95.5%]; adalimumab 133/140 [95.0%]).
At week 16, a significantly greater proportion of patients in the bimekizumab group achieved the primary endpoint of ACR50 compared with the placebo group (189/431 [44.0%] versus 28/281 [10.0%]; OR 7.1, 95% CI 4.5–10.9; p<0.0001; adalimumab: 64/140 [46.0%]). Moreover, the secondary endpoints were significantly greater in the bimekizumab group than placebo at week 16: the HAQ-DI score change from baseline (-0.26 versus -0.09; least squares [LS] mean difference -0.19, 95% CI -0.26, -0.13; p<0.0001; adalimumab: -0.33), Psoriasis Area and Severity Index (PASI90) response (133/217 [61.0%] versus 4/140 [3%]; OR 63.0, 95% CI 22.2–178.9; p<0.0001; adalimumab: 28/68 [41.0%]), SF-36 Physical Component Summary change from baseline (6.3 versus 2.3; LS means difference 4.3, 95% CI 3.2–5.4; p<0.0001; adalimumab: 6.8), Minimal Disease Activity response (194/431 [45.0%] versus 37/281 [13.0%]; OR 5.4, 95% CI 3.7–8.1; p<0.0001; adalimumab: 63/140 [45.0%]) and van der Heijde-modified total Sharp score change from baseline (0.01 [standard error (SsE) 0.04] versus 0.36 [SE 0.10]; LS means difference -0.33, 95% CI -0.52, -0.13; p=0.0012; adalimumab: -0.06 SE 0.08]). Furthermore, PASI100 response rates at week 16, observed as complete skin clearance, were higher in the bimekizumab group than the placebo group (103/217 [47.0%] versus 3/140 [2.0%], respectively). Patients receiving bimekizumab showed numerically improved scores compared with placebo for the Patients’ Assessment of Arthritis Pain (-23.6 [SE 1.3] versus -6.2 [SE 1.5]; adalimumab: -25.7 [SE 2.5]) and Functional Assessment of Chronic Illness Therapy–Fatigue (3.9 [SE 0.4] versus 1.5 [SE 0.5]; adalimumab: 5.0 [SE 0.7]).
At least one TEAE occurred in 258/431 patients (60.0%) in the bimekizumab group and 139/231 patients (49.0%) in the placebo group during the double-blind period (week 0–16) and in 300/431 patients (70.0%) in the bimekizumab group and 96/140 patients (69.0%) in the adalimumab reference group up to week 24 (weeks 0–24). Furthermore, treatment-emergent serious adverse events were recorded in seven patients (2.0%) in the bimekizumab group, three patients (1.0%) in the placebo group and two patients (1.0%) in the adalimumab reference group up to week 16. Eight patients (2.0%) in the bimekizumab group, three patients (1.0%) in the placebo group and three patients (2.0%) in the adalimumab group discontinued the study due to TEAEs. The most frequent TEAE in the bimekizumab group was nasopharyngitis (weeks 0–16: 40/431 [9.0%]; weeks 0–24: 50/431 [12.0%]). At week 16, one case of severe infection was reported in each of the bimekizumab and adalimumab groups, pneumonia and herpes zoster. Two additional serious infections (cellulitis and upper respiratory tract infection) and one severe infection (otitis media) occurred in the bimekizumab group and the adalimumab group, respectively.
By week 16, 20/431 (5.0%) patients in the bimekizumab group had fungal infections, including Candida infections (11/431 [3.0%]), and one (<1.0%) patient with moderate oral candidiasis discontinued the study. However, by week 24, fungal infections were experienced by 33/431 (8.0%) patients in the bimekizumab group, of whom 18 (4.0%) had Candida infections, and 15 (3.0%) had oral candidiasis. In the bimekizumab group, basal cell carcinoma and non-melanoma skin cancer occurred in one patient (<1.0%) by week 16 and week 24, respectively. Furthermore, one adjudicated major adverse cardiovascular event (myocardial infarction) that was not deemed treatment-related was recorded in a patient in the bimekizumab group. This patient had multiple cardiovascular event risks, including atherosclerosis, hypertension, abdominal aortic aneurysm and nicotine addiction.
In conclusion, the results from the BE OPTIMAL trial demonstrated the high efficacy and tolerability of bimekizumab in patients with PsA who are naïve to bDMARDs.
Bimekizumab in active psoriatic arthritis, previous inadequate response or intolerance to TNF-α inhibitors: BE COMPLETE, a phase III clinical trial
BE COMPLETE was a 52-week, multicentre, randomized, double-blind, placebo-controlled, phase III trial eligible for patients aged ≥18 years with adult-onset active PsA (≥6 months disease duration, TJC/SJC ≥3) and one or more active psoriatic lesions and/or a documented history of PSO.24 A total of 400 patients with a history of inadequate response or intolerance to one or more treatments with TNF-α inhibitors for PsA or PSO were included and randomized 2:1 to receive 160 mg subcutaneous bimekizumab (n=267) or placebo (n=133) every 4 weeks. Overall, 388 (97%) patients completed the trial to week 16, and 378 (95%) patients were enrolled at week 48 in the open-label extension study (160 mg bimekizumab every 4 weeks) currently in progress (BE VITAL; ClinicalTrials.gov identifier: NCT04009499).35 Of the patients, 306/400 (77%) had an inadequate response to one TNF-α inhibitor (TNF-i), whilst 45/400 (11%) had an inadequate response to two, and 49/400 (12%) had an intolerance to any TNF-α inhibitor. The primary endpoint was the proportion of patients in the bimekizumab group who had achieved ACR50 at week 16 compared with placebo.
At week 16, achieving an ACR50 response was significantly higher in the bimekizumab group compared with the placebo group (116/267 [43.0%] versus 9/133 [7.0%]; OR 11.1, 95% CI 5.4–23.0; p<0.0001). Additionally, the secondary endpoints were significantly greater at week 16 in the bimekizumab group compared with the placebo group: the HAQ-DI score (-0.38 versus -0.07; LS mean difference -0.33, 95% CI -0.42, -0.23; p<0.0001), PASI90 response (121/176 [69.0%] versus placebo 6/88 [7.0%]; OR 30.2, 95% CI 12.4–70.3; p<0.0001), SF-36 Physical Component Summary change from baseline (7.3 versus 1.4; OR 6.0, 95% CI 4.4–7.7; p<0.0001) and Minimal Disease Activity response (118/267 [44.0] versus 8/133 [6.0%]; OR 13.1, 95% CI 6.1–28.0; p<0.0001). Furthermore, the PASI100 response rate at week 16, observed as complete skin clearance, was 103/176 (59.0%) in the bimekizumab group and 4/88 (5.0%) in the placebo group. In the bimekizumab group, 59/176 (34.0%) achieved ACR50+PASI100 compared with 1/88 (1.0%) in the placebo group. Moreover, the Patients’ Assessment of Arthritis Pain (-27.7 [S.E 1.7] versus -4.5 [SE 2.1]) and Functional Assessment of Chronic Illness Therapy–Fatigue (5.5 [SE 0.6] versus 0.1 [SE 0.7) were numerically improved in the bimekizumab group compared with the placebo group.
At least one TEAE occurred in 108/267 patients (40.0%) in the bimekizumab group and 44/132 patients (33.0%) in the placebo group during the double-blind period (up to week 16). The most frequent TEAE in the bimekizumab group was nasopharyngitis (10/267 [4.0%]). Furthermore, TEAEs were recorded in five patients (2.0%) in the bimekizumab group and none in the placebo group. In the bimekizumab group, two patients with severe TEAEs discontinued treatment. In addition, two serious infections (bronchitis and coronavirus disease 2019 pneumonia) were reported. By week 16, 12/267 (4.0%) patients in the bimekizumab group had a fungal infection, of which seven (3.0%) were reported as Candida infections (all were oral candidiasis). However, no cases of severe fungal infections, cardiovascular events, uveitis, inflammatory bowel disease, suicidal ideation or behaviour, or malignancies (basal cell carcinoma) were reported. Six adjudicated major adverse hepatic events (increased liver enzyme levels) were recorded in the bimekizumab group; however, none led to discontinuation.
These results demonstrate the high efficacy and tolerability of bimekizumab in patients who were either inadequately responsive or intolerant to TNF-i. Furthermore, the therapeutic onset of bimekizumab was rapid, with a large difference in efficacy compared with the placebo at 4 weeks after the first administration, which may be valuable for patients who prioritize fast symptom relief. An open-label extension study (BE VITAL; ClinicalTrial.gov identifier: NCT04009499) is currently underway to evaluate the long-term efficacy and safety of bimekizumab.35
The resolution rate of enthesis and dactylitis: pooled BE OPTIMAL and BE COMPLETE data
The number of patients with entheses and/or dactylitis was lower than expected in the BE OPTIMAL and BE COMPLETED trials. In the pooled BE OPTIMAL and BE COMPLETE data, the resolution rates of enthesis and dactylitis were assessed using the Leeds Enthesitis Index and Leeds Dactylitis Index, respectively.23,24 Notably, the proportion of the complete resolution of enthesitis (bimekizumab 124/249 [50.0%] compared with placebo 37/106 [35.0%]; OR 1.9, 95% CI 1.2–3.1; p=0.0083; adalimumab 18/36 [50.0%]) and dactylitis (bimekizumab 68/90 [76.0%] compared with placebo 24/47 [51.0%], OR 3.4, 95.0% CI 1.6–7.6; p=0.0022; adalimumab 9/11 [82.0%]) was significantly higher in the bimekizumab group than in the other groups of the BE OPTIMAL trial.23
Bimekizumab in non-radiographic and radiographic axial spondyloarthritis: the phase III clinical trials BE MOBILE 1 and BE MOBILE 2
BE MOBILE 1 and BE MOBILE 2 were phase III, multicentre, randomized, double-blind, placebo-controlled trials conducted in parallel to investigate non-radiographic axial spondyloarthritis (nr-axSpA) and radiographic axSpA (r-axSpA), respectively, in individuals aged ≥18 years with active axSpA ( Bath Ankylosing Spondylitis Disease Activity Index ≥4 and spinal pain ≥4).36
In BE MOBILE 1, nr-axSpA was determined by clinical diagnosis and by fulfilling the Assessment of SpondyloArthritis International Society (ASAS) classification criteria in addition to objective inflammation (active sacroiliitis on magnetic resonance imaging) and/or elevated C-reactive protein (≥6.0 mg/L). In BE MOBILE 2, patients had r-axSpA and fulfilled the modified New York criteria, including radiographic evidence of sacroiliitis (grade ≥2 bilateral or grade ≥3 unilateral). In BE MOBILE 1, patients (n=254) were randomized 1:1 to receive 160 mg subcutaneous bimekizumab (n=128) or placebo (n=126) every 4 weeks, and 244 (96.1%) patients completed the double-blind treatment period to week 16. In contrast, in BE MOBILE 2, patients (n=332) were randomized 2:1 to receive 160 mg subcutaneous bimekizumab (n=221) or placebo (n=111) every 4 weeks, and 313 (94.3%) patients completed to week 16.
The primary endpoint was met in both studies, as ASAS40 response was significantly higher in the bimekizumab group compared with the placebo group (nr-axSpA: 61/128 [47.7%] versus 27/126 [21.4%], p<0.001; r-axSpA: 99/221 [44.8%] versus 25/111 [22.5%], p<0.001). Furthermore, disease activity (Bath Ankylosing Spondylitis Disease Activity Index and Ankylosing Spondylitis Disease Activity Score), physical function (Bath Ankylosing Spondylitis Functional Index and SF-36 Physical Component Summary), pain (nocturnal spinal pain), quality of life (Ankylosing Spondylitis Quality of Life) and spinal mobility (Bath Ankylosing Spondylitis Metrology Index) improved in the bimekizumab group. Through weeks 0 to 24, ≥1 TEAE occurred in 124/244 (50.8%) patients with nr-axSpA and 183/330 (55.5%) patients with r-axSpA. TEAEs leading to trial discontinuation occurred in 3/244 (1.2%) patients with nr-axSpA and 12/330 (3.6%) patients with r-axSpA. Notably, the most frequently reported TEAEs were nasopharyngitis, upper respiratory tract infection and pharyngitis. In addition, fungal infections were reported in 9/128 (7.0%) patients with nr-axSpA and 13/221 (5.9%) patients with r-axSpA. Oral candidiasis was reported in 4/128 (3.1%) patients with nr-axSpA and 9/221 (3.4%) patients with r-axSpA. Through these results, the efficacy and tolerability of bimekizumab in patients with nr-axSpA and r-axSpA were demonstrated.
Discussion and conclusions
In this article, we reviewed the high efficacy and tolerability of bimekizumab in patients with active PsA from two phase III clinical trials, BE ACTIVE and BE OPTIMAL.23,24 Meta-analyses have already demonstrated that the efficacy of IL-17A, IL-17RA-i and IL-23-i is comparable to anti-TNFs for arthritis (ACR50 response rate: IL-17A-i 27.0–31.2%; IL-17RA-i 24.6%; IL-23-i 27.1%; TNF-i 27.6–52.9%) and preferable efficacy for skin manifestations (PASI 90 response rate: IL-17A-i 35.0–44.9%; IL-17RA-i 52.7%; IL-23-i 60.3%; TNF-i 13.5–27.0%), enthesitis (resolution of enthesitis; IL-17A-i 33.1–44.4%; IL-17RA-i 38.0%; IL-23-i 42.3%; TNF-i 39.7%) and dactylitis (resolution of dactylitis: IL-17A-i 50.5–62.4%; IL-17RA-i 53.6%; IL-23-i 60.4%; TNF-i 51.3%).37 These two studies have shown that bimekizumab demonstrates superior efficacy over other existing drugs (not based on head-to-head clinical trials) in several ways. First, the PASI clearance rate was higher (≥50.0%). Second, the ACR20 and ACR50 (a stricter primary endpoint in the bimekizumab studies) achievement rates were higher in both naïve and inadequate response or intolerance to TNF-i. Third, complete enthesitis and dactylitis resolutions were comparable to those of other existing drugs (i.e. IL-17A, TNF-i). Fourth, the observation period was short (16 weeks). Furthermore, a recent report demonstrated the efficacy of bimekizumab in patients with moderate-to-severe plaque PSO who showed an inadequate response or intolerance to IL-17-i.38 This efficacy may be due to the difference in IL-17A inhibition and dual IL-17A/IL-17F inhibition; therefore, such an effect is also expected for PsA. However, a trial assessing the efficacy and tolerability of bimekizumab in patients with PsA who show an inadequate response or are intolerant to IL-17A inhibitors has yet to be performed. Additionally, it is unclear how the discrepancy in efficacy between bimekizumab, which showed numerically superior results, and brodalumab, which possesses broader IL-17 family signal-blocking activity, can be explained.39
The emergence rate of candidiasis tended to be numerically higher in bimekizumab (plaque PSO ≥10.0%, PsA~3.o%) than in IL-17A (PsA ~1.0%), and its trend was significant in patients with plaque PSO who received 320 mg of bimekizumab. This finding may be attributed to the dose and the differences between IL-17A inhibition and dual IL-17A/IL-17F inhibition.
Considering the rapid and higher efficacy of bimekizumab in skin and arthritis than other existing IL-17A-i and IL-17-RA blockades, dual IL-17A/IL-17F inhibition and inhibition of signals via IL-17RA/RC by bimekizumab could be a novel blockbuster to overcome the unmet needs of PsA. However, this unique mode of action may also contribute to a higher risk of candidiasis.
Although its real-world, long-term efficacy and safety profile need to be well validated, bimekizumab is expected to provide novel, insightful knowledge that might lead to better patient outcomes and a more precise understanding of the pathogenesis of PsA (i.e. a more detailed role of IL-17A, IL-17F and their receptors).