Psoriatic arthritis (PsA) is a chronic inflammatory disease with an estimated global prevalence of 0.2%.1,2 The condition may occur in up to 30% of patients with psoriasis, with PsA typically developing 10 years after the onset of skin disease.1,3,4 PsA is a highly heterogeneous disease, with manifestations including peripheral arthritis, axial disease, psoriasis, nail changes, dactylitis and enthesitis, alongside extra-articular manifestations such as uveitis and inflammatory bowel disease.4 In addition, PsA has a significant impact on the patient’s physical function and quality of life, and is associated with an increased prevalence of cardiovascular comorbidity, depression and fibromyalgia.5–7 As a result, it poses a significant financial burden to both the individual and society.8 Greater understanding of the immunopathogenesis of PsA has allowed its treatment to evolve rapidly over the past two decades with the introduction of biologic agents to improve the varied features of the disease. In this review, we focus specifically on the benefits of inhibiting interleukin-23 (IL-23) for PsA, with the emphasis on monoclonal antibodies targeting the p19 subunit of IL-23.
Pathophysiology and the role of interleukin-23 in psoriatic arthritis
The pathogenesis of PsA results from dysregulation of the immune system, triggered by both genetic and environmental factors and resulting in the proliferation of immune cells and consequent inflammation.9 Genetic susceptibility plays an important role in the development of both psoriasis and PsA. First-degree relatives have a risk ratio of 40 for the development of PsA.10 Numerous human leukocyte antigen (HLA) alleles, including HLA-B27, have been identified as being associated with susceptibility to the development of PsA, as well as certain disease manifestations and latency between the skin and joint features.11,12 Non-HLA genetic factors have also been identified, including polymorphisms in the gene encoding the IL-23 receptor.13 Additionally, various environmental risk factors have been associated with the development of both psoriasis and PsA, including obesity and musculoskeletal injury.14,15
The IL-23/IL-17 pathway is critical in the development of PsA, as well as in psoriasis and other spondyloarthropathies.16,17 IL-23 represents an upstream regulatory cytokine composed of a p19 subunit, which is unique to IL-23, and a p40 subunit that is shared with IL-12. IL-23 binds to its receptor, modulating the expansion of CD4+ Th17 cells with the involvement of Janus kinase (JAK) signalling pathways, resulting in increased expression of downstream cytokines, including IL-17A, tumour necrosis factor (TNF) and IL-22.17,18 Via its downstream signalling effects, IL-23 plays a key proinflammatory role in the skin, synovium and enthesis.19 Dysregulation and overexpression of IL-23 are core to the pathophysiology of PsA and other spondyloarthropathies.18 Unlike rheumatoid arthritis, where the joint pathology primarily involves the synovium, enthesitis is thought to be the fundamental process in spondyloarthropathies, with synovitis occurring secondarily.20 IL-23 acts directly at the enthesis. Animal studies have shown that entheseal T cells respond to IL-23 in vitro triggering other inflammatory mediators, while human studies have demonstrated that myeloid cells located within the enthesis produce IL-23 and other inducible cytokines that mediate disease.21,22 Due to this pivotal role in promoting inflammation in PsA, IL-23 has become a major target for treatments aimed at improving the many features of the condition.
Current guidelines for the treatment of psoriatic arthritis
Pharmacological options for the management of PsA include non-steroidal anti-inflammatory drugs (NSAIDs); conventional, synthetic, disease-modifying, anti-rheumatic drugs (csDMARDs); biological DMARDs (bDMARDs), including TNF inhibitors, IL-17 inhibitors, IL-12/23 inhibitors, selective IL-23 inhibitors and abatacept; JAK inhibitors; and the phosphodiesterase-4 (PDE-4) inhibitor apremilast.23 Treatment choice is determined by disease activity, including severity of skin involvement or presence of axial disease, comorbidities, patient preference, and frequency of administration, as well as local prescribing guidelines and healthcare remuneration.
A stepwise approach to the treatment of PsA is generally recommended. Guidelines suggest that NSAIDs may be considered as an initial symptomatic treatment for PsA, with more limited recommendations on the role for local or systemic glucocorticoids.23–25 In the setting of poor prognostic factors, csDMARDs should be considered earlier in the management paradigm.24 Methotrexate is generally recommended as the csDMARD of choice, particularly for patients with psoriasis, in whom it has proven effective.24 Other csDMARD options include leflunomide and sulfasalazine. Of note, the American College of Rheumatology (ACR) recommends that TNF inhibitors should be commenced as first-line therapy rather than csDMARDs in treatment-naive patients with active PsA, particularly in severe PsA or severe psoriasis.23 If treatment targets are not achieved with these strategies, a bDMARD targeting either TNF, IL-17, IL-12/23 or IL-23 should be initiated, with particular consideration given to the predominant disease manifestations, with all of these bDMARDs being suitable in the setting of peripheral arthritis, enthesitis or dactylitis.25 If axial disease predominates, a TNF or IL-17 inhibitor is typically preferred as first-line therapy, whereas an IL-23, IL-12/23 or IL-17 inhibitor may be preferred in the setting of significant psoriasis.24,25 Additionally, IL-17 inhibitors and the TNF inhibitor etanercept should be avoided in the setting of concomitant inflammatory bowel disease. The use of JAK inhibitors has generally been reserved for initial bDMARD failure, although this is changing, with the latest recommendations from the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) suggesting they can be used as first-line agents for multiple domains of PsA.24,25 PDE-4 inhibition with apremilast is an option for patients who have milder disease with inadequate response to csDMARDs but contraindications to other agents.24 Critically, it should be noted that both the current European League Against Rheumatism and ACR guidelines pre-date the studies and availability of selective IL-23 inhibitors for PsA, whereas these agents have been included in the recently updated GRAPPA recommendations.
A treat-to-target approach is also recommended, although this can be more complex than in conditions such as rheumatoid arthritis, given the varied disease manifestations of PsA.26 Multiple disease activity scores exist for PsA, with the ACR 20% response criteria (ACR20) typically used as the primary outcome measure for studies on peripheral arthritis. The Psoriasis Area and Severity Index (PASI) is used to assess psoriasis, while composite scores, such as the Minimal Disease Activity (MDA), have been developed to assess disease activity across multiple domains.27
Methods
A literature search of PubMed was conducted using the search terms ‘psoriatic arthritis’ alongside ‘guselkumab’, ‘risankizumab’, ‘tildrakizumab’ and ‘ustekinumab’ up to September 2021. The included studies were limited to randomized controlled trials and open-label extension studies, including abstract data. Selective studies from the psoriasis data were also selected. A broader literature search using PubMed and the references of the selected articles was undertaken to explore the background literature on PsA to support this review.
Efficacy of interleukin-23 inhibition in psoriatic arthritis
Guselkumab, risankizumab and tildrakizumab are all selective IL-23 inhibitors that bind to the p19 subunit of IL-23 and are approved for use in psoriasis, with growing evidence regarding their role in PsA. Mirikizumab, also a selective IL-23 inhibitor, has been studied in psoriasis and inflammatory bowel disease, but is yet to be investigated in the treatment of PsA.28,29 Ustekinumab, an IL-12/23 inhibitor, is approved for use in both psoriasis and PsA, and is included in this review due to its effect on IL-23 inhibition.
Guselkumab
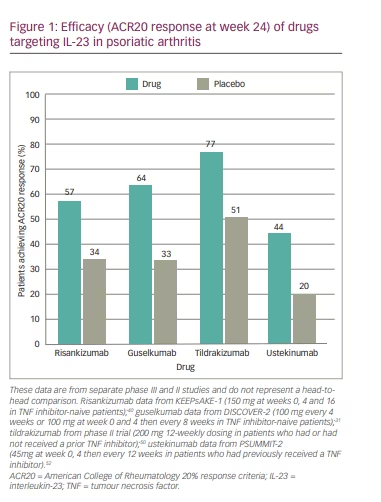
Guselkumab is a fully human monoclonal antibody that binds the p19 subunit of IL-23, and was the first selective IL-23 inhibitor to be approved for use in PsA in the USA.30 The DISCOVER-1 and -2 phase III trials randomly assigned patients to receive either guselkumab (100mg every 4 weeks or 100 mg at weeks 0 and 4, then every 8 weeks) or placebo.31,32 The primary endpoint of ACR20 at week 24 was achieved by a significantly greater proportion of patients in both guselkumab groups (59–64% in the 4-weekly group and 52–64% in the 8-weekly group) compared with the placebo group (22–33%).31,32 Consistent treatment responses were seen in patients who received methotrexate at baseline and in those who had a previous inadequate response to TNF inhibitors.31 Compared to placebo, ACR50 response was higher in both guselkumab dose groups and ACR70 response was significantly greater in all guselkumab groups except the 8-weekly group in DISCOVER-1. This was similar for radiographic progression, where significant decreases were only demonstrated in the 4-weekly group. Improvements in the Disease Activity Index Score in 28 joints calculated with C-reactive protein (DAS28-CRP), PASI response, dactylitis and enthesitis, and functional and quality-of-life scores were also demonstrated.31,32 Improvements across multiple domains were sustained at week 52 both in patients who were biologic-naive and those who had previously received TNF inhibitors.33–35 Additionally, post hoc analysis showed greater improvements in scores for axial disease in patients with associated imaging-confirmed sacroiliitis.36
In psoriasis, guselkumab performed better than the TNF inhibitor adalimumab, had superior long-term efficacy compared with the IL-17 inhibitor secukinumab and demonstrated efficacy in patients with previous inadequate response to ustekinumab.37–39
Risankizumab
Risankizumab has been approved for use in moderate-to-severe psoriasis and had just received Food and Drug Administration (FDA) approval for PsA at the time this article was published.40 The pivotal KEEPsAKE-1 phase III study examined the efficacy of risankizumab compared with placebo in patients with active PsA who had inadequate response or intolerance to at least one csDMARD.41 In the complementary KEEPsAKE-2 study, risankizumab was compared with placebo in active PsA with inadequate response or intolerance to one or two bDMARDs or at least one csDMARD.42 Active PsA was defined as having at least five swollen and tender joints. In KEEPsAKE-1, 964 patients were randomly assigned to receive either risankizumab 150 mg or placebo at weeks 0, 4 and 16. The primary endpoint of ACR20 at week 24 was achieved in a significantly greater proportion of patients in the risankizumab group (57.3% versus 33.5% in the placebo group).41 In KEEPsAKE-2, a total of 443 patients were assigned to the same treatment groups, with a significantly greater number meeting the primary endpoint of ACR20 in the risankizumab group than in the placebo group (51.3% versus 26.5%, respectively).42 In both studies, significant improvements in multiple secondary endpoints were also demonstrated, including ACR20 at week 16, ACR50 and PASI 90 at week 24, MDA, functional and quality-of-life scores, and resolution of enthesitis and dactylitis. The latest available long-term data from the open-label extension period showed sustained response through 52 weeks, with ACR20/50/70 attained in 70%, 43% and 26% of patients receiving risankizumab, respectively, in KEEPsAKE-1 and in 58%, 32% and 17% of patients, respectively, in KEEPsAKE-2.43 Risankizumab was well tolerated, with a safety profile consistent with that established in the treatment of moderate-to-severe psoriasis.41,42 Earlier phase II studies also revealed evidence for inhibition of radiographic progression.44
There are strong data supporting the role of risankizumab in the management of psoriasis alone, with studies demonstrating superior efficacy to adalimumab and ustekinumab, and superior long-term efficacy compared with secukinumab.45–47 The efficacy of risankizumab for axial disease in PsA is unclear, with data from patients with ankylosing spondylitis failing to show significant clinical improvements with risankizumab compared with placebo.48 Post hoc analysis in relation to this from the KEEPsAKE trials is pending. Risankizumab has also shown promise for the treatment of inflammatory bowel disease in phase II studies.49
Tildrakizumab
Tildrakizumab is currently approved for the treatment of severe psoriasis.50 To date, one phase II study has evaluated the efficacy and safety of tildrakizumab for the treatment of PsA.51 Patients were randomized to receive tildrakizumab 200 mg 4-weekly, 200 mg 12-weekly, 100 mg 12-weekly, 20 mg 12-weekly or placebo, with the latter two groups being switched to tildrakizumab 200 mg 12-weekly at week 24. The primary endpoint of ACR20 at week 24 was achieved by a significantly higher proportion of patients receiving any dose of tildrakizumab (71–80% versus 51% receiving placebo); however, this was not significant in those who had previously received a TNF inhibitor. PASI response rates were also higher. Patients receiving 200 mg doses achieved higher rates of ACR50/70, DAS28-CRP, MDA, and physician and patient assessment scores of disease activity; however, improvement in dactylitis and enthesitis was not demonstrated.
Ustekinumab
Ustekinumab is a human immunoglobulin G1 monoclonal antibody that binds to the shared p40 subunit of IL-12 and IL-23, thus inhibiting the differentiation and activation of both Th1 and Th17 cells and, consequently, several key cytokines, such as IL-23, IL-17 and TNF.9 It has been approved for use in PsA since 2013. The PSUMMMIT-1 and -2 phase III trials randomly assigned a total of 927 adults with active PsA to either placebo, ustekinumab 45 mg or ustekinumab 90 mg at 0 and 4 weeks, then every 12 weeks.52,53 The primary endpoint of ACR20 at 24 weeks was achieved by 42–44% of patients who received 45 mg and by 46–50% who received 90 mg, with responses being significantly greater than placebo (20–23%) in both groups and being maintained at 52 weeks. Significant differences were also found for the ACR50 and ACR70 responses. Improvements were also seen in dactylitis, psoriasis, DAS28-CRP, Bath Ankylosing Spondylitis Disease Activity Index and Health Assessment Questionnaire Disability Index scores, and radiographic progression. Efficacy was also observed in patients who had previously received a TNF inhibitor.52–54
Ustekinumab has demonstrated greater efficacy than the TNF inhibitor etanercept for skin psoriasis.55 However, it failed to show benefit in both radiographic and non-radiographic axial spondyloarthritis.56
Key efficacy data for all four agents are shown in Figure 1.
Safety summary
One of the major benefits of both selective IL-23 and IL-12/13 inhibitors is their tolerability. The frequency of serious adverse events, such as infection, malignancy and major adverse cardiac events (MACE), has been consistently similar to placebo in studies of psoriasis where longer-term data are available.57 Safety data from trials of selective IL-23 inhibitors in PsA support this low incidence of serious adverse events.33,42 Common side effects include upper respiratory tract infections, headaches and injection-site reactions, and discontinuation rates have been low.33,42,57 Rates of serious infections are rare, with no signal to suggest increased risk of tuberculosis infection or reactivation; however, longer-term evidence is required.33,42,57 Current data do not suggest that IL-23 inhibitors increase the risk of COVID-19 disease or of poorer outcomes.58 There remains a theoretical concern of increased risk of infection; thus, screening for tuberculosis, hepatitis B and C, and HIV prior to commencement is generally performed, as is routine for other biologics.59 Infrequent MACE have been reported in clinical trials of selective IL-23 inhibitors; however, the rates were in line with that expected in the general population, with cases typically having identifiable cardiovascular risk factors.57 Interestingly, briakinumab, another IL-12/23 inhibitor, had its FDA approval withdrawn due to a reported increase in MACE, although trials with ustekinumab have not consistently found increased risk compared with placebo.60,61 There is currently a lack of safety data to guide consensus recommendations on the use of IL-23 inhibitors in pregnant women with either psoriasis or PsA. Limited data suggest that IL-12/23 inhibitors are not associated with significantly increased risk of negative maternal-foetal outcomes; however, clinical experience with selective IL-23 inhibitors in this area is even more limited.62
Discussion
IL-23 inhibitors are now well established as an effective treatment for psoriasis and have a growing body of evidence to support their use in PsA.31,32,41,42 The IL-23p19 inhibitor risankizumab has demonstrated improvements in disease outcomes across multiple domains of PsA, similar to those seen for guselkumab.41,42 The efficacy of IL-23 inhibitors appears to differ depending on the site of disease, with particularly impressive improvements or even complete resolution of skin psoriasis.37-39, 45-47 Efficacy in peripheral arthritis, enthesitis and dactylitis has also been demonstrated.31,32,41,42 This has been seen both in patients who are biologic-naive and in those who have had an inadequate response to TNF inhibition. In addition, data from open-label extension periods have confirmed the sustained response of selective IL-23 inhibition in both PsA and psoriasis.33,34
The role of IL-23 inhibition in patients with psoriatic spondyloarthritis is less clear. The prevalence of axial disease in PsA ranges from 25% to 70%, depending on the definition.63 Both ustekinumab and risankizumab failed to demonstrate efficacy in ankylosing spondylitis.56,48 However, earlier studies of ustekinumab looking at axial involvement in PsA did suggest benefit in axial disease,64 and data from the DISCOVER trials showed improvement in imaging-confirmed sacroiliitis in patients who received guselkumab.35 Interestingly, given the common IL-17/23 pathway, IL-17 inhibitors have proven benefit in axial disease in both PsA and ankylosing spondylitis.65,66 This unclear responsiveness of axial disease to IL-23 inhibition suggests that IL-23 may play less of a role in the pathogenesis of inflammation in the axial skeleton, with the contribution of IL-23 being primarily through IL-17 upregulation, and that the interaction between IL-17 and IL-23 may be tissue specific.67
The differential effects of IL-23 and IL-17 inhibition at distinct tissue sites is further supported by outcomes of studies in inflammatory bowel disease. Ustekinumab has proven efficacy for the treatment of both Crohn’s disease and ulcerative colitis.68,69 Studies of the IL-23p19 inhibitors are also promising.29,70,71 Conversely, IL-17 inhibitors should be used with caution in patients with PsA and coexistent inflammatory bowel disease due to the possible risk of flare, as well as lack of efficacy for bowel disease.72 Selective IL-23 inhibitors, ustekinumab and IL-17 inhibitors all have outstanding efficacy for skin psoriasis, being superior to earlier biologics, namely TNF inhibitors.45,55,73 Notably, risankizumab and other selective IL-23 inhibitors have demonstrated superior efficacy to both ustekinumab and IL-17 inhibitors.46,47 There are currently no head-to-head data comparing IL-23 and IL-17 inhibition in PsA, with both drug targets demonstrating improvements in ACR20 responses alongside multiple secondary outcome measures.74 It is worth noting that in the head-to-head EXCEED trial, secukinumab did not meet statistical significance for superiority versus adalimumab in the primary endpoint of ACR20 response.75 Improving our understanding of the precise role of the IL-23/IL-17 axis in the varied phenotypes of PsA is key to delivering precision medicine in this area.
When comparing the selective IL-23 inhibitors and the IL-12/23 inhibitor ustekinumab, both were found to have efficacy in similar PsA disease domains, namely psoriasis, peripheral arthritis, dactylitis and enthesitis, as well as inflammatory bowel disease. While IL-12/23 inhibitors have demonstrated efficacy in axial disease, further data are required for selective IL-23 inhibitors. It has been postulated that it is the inhibition of IL-23 rather than IL-12 that underlies the efficacy of ustekinumab in psoriasis, and this is possibly also applicable to PsA.75
Given the expanding range of biologic and targeted, synthetic, disease-modifying agents available for use in PsA, further head-to-head trials are needed to improve our understanding of where IL-23 inhibitors fit in our treatment paradigm. The recently updated GRAPPA guidelines recommend use of drugs targeting the p19 subunit of IL-23 in multiple disease domains, including peripheral arthritis, enthesitis, dactylitis and psoriasis.25 Older guidelines suggest preference of an IL-12/23 or IL-17 inhibitor in the setting of PsA with severe psoriasis.23,24 Given their superior results in psoriasis and comparable results in peripheral arthritis, selective IL-23 inhibitiors may become the preferential agent in such patients. These agents have a favourable risk profile, which may make them a preferred biologic agent in certain patient groups, although we await longer-term safety data. At present, the drug selection process of clinicians is currently guided by clinical phenotype, personal experience, patient preferences and previously trialled therapies. Improved understanding of the immune pathways in PsA and the establishment of predictive biomarkers will aid clinicians in moving towards precision medicine.
Conclusion
The therapeutic repertoire for the management of PsA is expanding rapidly, with inhibition of IL-23 being pivotal in this process. IL-23p19 inhibitors are establishing themselves as an effective and well-tolerated agent in the treatment of PsA, with strong phase III data and longer-term extension studies. The future is certainly bright for individuals suffering from PsA, although individualized intervention remains elusive. Further head-to-head data are required to determine where IL-23 inhibitors best fit within the treatment paradigm of PsA.