Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) includes several distinct clinical entities: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA) and renal-limited vasculitis. What all these diseases have in common is the presence of serum autoantibodies, designated as ANCA, that react with antigens in the cytoplasm of neutrophilic granulocytes and monocytes.1,2
The GPA type, formerly known as Wegener’s granulomatosis3 is a rare form of multisystemic vasculitis affecting predominantly small-to-medium vessels.4 GPA is a necrotizing granulomatous inflammation and vasculitis involving the upper and lower respiratory tract and is frequently associated with necrotizing pauci-immune glomerulonephritis. ANCA in GPA most commonly targets the proteinase 3 antigen (PR3) and gives the cytoplasmic (c-ANCA) pattern of staining on ethanol-fixed neutrophils used for ANCA detection. MPA, in contrast, presents histologically as a vasculitis lacking typical granulomatous inflammation and frequently causing rapidly progressive pauci-immune glomerulonephritis and life-threatening alveolar haemorrhage. MPA is commonly associated with ANCA, giving a characteristic perinuclear staining pattern and primarily targeting the enzyme myeloperoxidase (MPO).5 The final systemic form of AAV, EGPA (also known as Churg–Strauss syndrome), in addition to features of systemic vasculitis, is characterized by the presence of blood and tissue eosinophilia in patients with a history of adult-onset asthma. Interestingly, only about 40% of patients with EGPA have detectable ANCA antibodies, which are mostly reactive with the MPO antigen.6
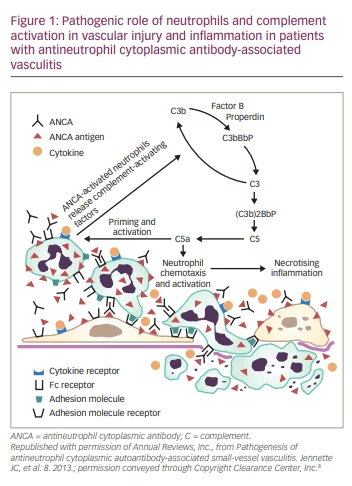
Role of complement activation in the pathogenesis of ANCA-associated vasculitis
Interactions between environmental (infections, drugs, silica exposure) and host factors can lead to a breach in tolerance, initiating the production of pathogenic ANCAs in genetically susceptible individuals.7 Once produced, these antibodies in turn can find their autoantigen targets (PR3 and MPO antigens) expressed on the surface of (cytokine)-primed neutrophils, which subsequently may initiate complement activation. This process depends on both Fc receptor engagement and F(ab)2 crosslinking of ANCA antibodies on the surface of neutrophils. One of the most important complement fragments generated by complement activation is complement (C)5a, which upon binding to the C5a receptor (C5aR), amplifies the inflammation loop by not only acting as a potent neutrophil chemoattractant but also causing vascular injury through neutrophil degranulation, with subsequent release of lysosomal proteases and oxygen free radicals. Consequently, this may result in extensive tissue damage and increased vascular permeability, ultimately causing inflammatory necrotizing vasculitis (Figure 1). AAV is known to go through frequent relapses, which can further aggravate tissue injury leading to permanent organ damage and diminished health-related quality of life.
Evidence for the important role of complement in the pathogenesis of AAV comes primarily from animal studies.9–11 Transfer of anti-MPO immunoglobulin (Ig)G antibodies into healthy mice or transfer of anti-MPO spleen cells into immune-deficient mice can cause crescentic glomerulonephritis in a complement-dependent manner by activating the alternative complement pathway. Support for this hypothesis came from experiments in mice lacking factor B of the alternative pathway, which remained healthy.12 Similarly, mice lacking C5 did not develop the disease in contrast to mice deficient in C4 from the classical pathway of activation, which developed crescentic glomerulonephritis.12 Human in vitro culture experiments showed that incubation of neutrophils with either MPO-ANCA or PR3-ANCA antibodies, but not with control immunoglobulins, caused release of factors that could activate the complement system.13 Additional support for the role of complement in AAV in humans came from studies showing the presence of activated components of the alternative complement pathway in blood and inflamed renal tissue of patients with AAV.14,15
Oral administration of a small-molecular antagonist of human C5aR/CD88 resulted in blockage of anti-MPO-induced necrotizing and crescentic glomerulonephritis in mice expressing human C5aR/CD88, which was the first hint that blockage of C5aR might provide therapeutic benefits to patients with AAV.16
Current standard of treatment for ANCA-associated vasculitis
Prior to the introduction of effective therapy, patients with GPA had an extremely grave prognosis and high mortality rate (82% mortality in 1 year, with median survival of about 5 months).17 The introduction of therapy with the alkylating agent cyclophosphamide, in combination with glucocorticoids, greatly improved patient outcomes.13,18 This treatment remained the mainstay of therapy for five decades despite having a relatively high toxicity profile. It was only in 2011 that a monoclonal antibody targeting the CD20 molecule on the surface of B cells, rituximab, received US Food and Drug Administration (FDA) approval for the treatment of GPA.19 Clinical trials have demonstrated clearly that treatment with rituximab is noninferior to cyclophosphamide.20,21 The rituximab in ANCA-associated vasculitis (RAVE) trial (ClinicalTrials.gov identifier: NCT00104299) showed rituximab to be superior to cyclophosphamide for inducing remission in relapsing AAV, with induction of remission in 34 of 51 patients (67%) in the rituximab group compared with 21 of 50 patients (42%) in the cyclophosphamide group (p=0.01).20 Use of rituximab avoids the risk of infertility, haematological malignancies and bladder toxicity that can be seen with use of cyclophosphamide. Furthermore, rituximab is the preferred agent for the maintenance of remission when compared with methotrexate or azathioprine.22,23 In 2018, the FDA extended the rituximab label indication for the maintenance of remission in patients with AAV.24 This clearly signifies the importance of B-cells in the immunopathogenesis of GPA through their role as antigen-presenting/proinflammatory cytokine-secreting cells and precursors of autoreactive ANCA-secreting plasma cells. Interestingly, some B-cell subsets may also finely tune an on-going immune response through their ability to secrete interleukin (IL)-10 and other regulatory cytokines,25 leading us to wonder whether their function may be insufficient in patients with AAV.
Rituximab and other B-cell-targeted therapies in the pipeline,26,27 however, are not exempt from life-threatening side effects, such as progressive multifocal leukoencephalopathy, although this is extremely rare and mostly seen among patients with haematological malignancies.28 Therefore, the goal of on-going research is to identify novel therapeutics that could provide better overall control of necrotizing inflammation without causing dangerous immunosuppression. We are looking into agent(s) that would prove to be efficient not only as early induction agents, but also as preferred agents for the maintenance of remission, allowing relatively fast prednisone taper while minimizing the risk of possible relapse or refractory disease activity. Optimal therapy for GPA remains to be determined, as no current regimen meets all these expectations. High mortality rates still range between 10% and 15% in the first year after starting conventional treatment with immunosuppressive medications and glucocorticoids.29 This remains of high concern, as approximately 50% of all mortalities can be attributed to treatment-related infectious complications.29
Based on these limitations, one important disease strategy is to minimize glucocorticoid exposure given that glucocorticoids have been an integral part of historical and current therapies for GPA. With patients being subjected to extended courses of glucocorticoid therapy, the associated glucocorticoid-related morbidity has been to the patients’ detriment. The plasma exchange and glucocorticoids for treatment of ANCA-associated vasculitis (PEXIVAS; ClinicalTrials.gov identifier: NCT00987389) study shed light on potential benefits of a reduced-dose glucocorticoid regimen, which was noninferior to the standard high-dose glucocorticoid therapy, specifically in mortality and end-stage renal disease and was associated with fewer serious infections at the 1-year landmark.30 The cumulative dose of oral glucocorticoids in the accelerated-prednisone taper group was 60% lower at 6 months compared to the standard prednisone taper group.30 Pepper et al. further studied the effect of rapid glucocorticoid tapering over a period of 1–2 weeks in 49 patients with AAV and showed reduced incidence of infections and diabetes, while maintaining the study efficacy compared to the standard prednisone taper group.31 Therefore, a step forward would be to identify effective glucocorticoid-sparing regimens with similar treatment outcomes and safety profile.
Potential glucocorticoid-sparing and disease-modifying effects of avacopan in ANCA-associated vasculitis: results from the recent phase III clinical trial
Jayne et al. investigated whether an oral small-molecular antagonist of C5aR, avacopan (Tavneos®; CCX168; ChemoCentryx, Inc., San Carlos, CA, USA), could be used as an adjuvant to rituximab and cyclophosphamide in the induction of remission for AAV in the ADVOCATE trial (ClinicalTrials.gov identifier: NCT02994927).33 As already elaborated above, activation of the alternative complement pathway has been implicated in the pathogenesis of AAV and this has been corroborated in a murine model of AAV,16 as well as in the phase II CLEAR trial (ClinicalTrials.gov identifier: NCT01363388).34 In the CLEAR trial, patients with AAV were initially treated with the standard induction regimen, either rituximab or cyclophosphamide, and then randomly assigned to one of the treatment groups: avacopan only, avacopan with low-dose prednisone, or control group treated with high-dose prednisone alone. Both avacopan groups were noninferior to the control group.34 Furthermore, patients on avacopan showed lower urinary albumin levels and had a better quality of life.34
In the ADVOCATE phase III clinical trial, 143 centres participated in this double-blind, randomized, double-dummy controlled trial that recruited a total of 331 patients.33 The trial included patients with either newly diagnosed or relapsing GPA and MPA in whom treatment with either cyclophosphamide or rituximab was indicated. Patients had to be older than 18 years, test positive for either PR3 or MPO antibodies and have an estimated glomerular filtration rate (eGFR) of >15 mL/min/1.73 m2. Additionally, patients had to have at least one major item or at least three non-major items, or at least two renal items (haematuria and proteinuria) on the Birmingham Vasculitis Activity Score (BVAS). Patients were excluded if they had any of the following criteria: received more than 3 g intravenous (IV) glucocorticoids within 4 weeks, or >10 mg/day oral prednisone for more than 6 weeks continuously before screening; diffuse alveolar haemorrhage requiring mechanical ventilation anticipated to last beyond screening; other multisystemic autoimmune disease, coagulopathy or a bleeding disorder; undergoing dialysis or plasma exchange; received cyclophosphamide treatment 12 weeks prior to screening; received rituximab or other B-cell antibody within 52 weeks of screening or 26 weeks provided B-cell reconstitution had occurred (i.e. CD19 count >0.01×109/L); received anti-tumour necrosis factor treatment, abatacept, alemtuzumab, IVIg, belimumab, tocilizumab or eculizumab within 12 weeks prior to screening; or had a kidney transplant.
Randomization and treatment
Of the 368 patients assessed for eligibility, 331 underwent randomization. Similar patient numbers were assigned to the avacopan group (166 assigned, 166 received avacopan) and prednisone group (165 assigned, 164 received prednisone). All patients, regardless of the assigned treatment group, were exposed to one of three induction regimens, which was left to the discretion of the provider: (1) IV cyclophosphamide 15 mg/kg (up to 1.2 g) on day 1 and at weeks 2, 4, 7, 10 and 13, followed by oral azathioprine (target dose of 2 mg/kg per day); (2) oral cyclophosphamide 2 mg/kg (up to 200 mg) per day for 14 weeks, followed by oral azathioprine (target dose 2 mg/kg/day); or (3) IV rituximab 375 mg/m2 of body surface area per week for the total of 4 weeks (Figure 2).
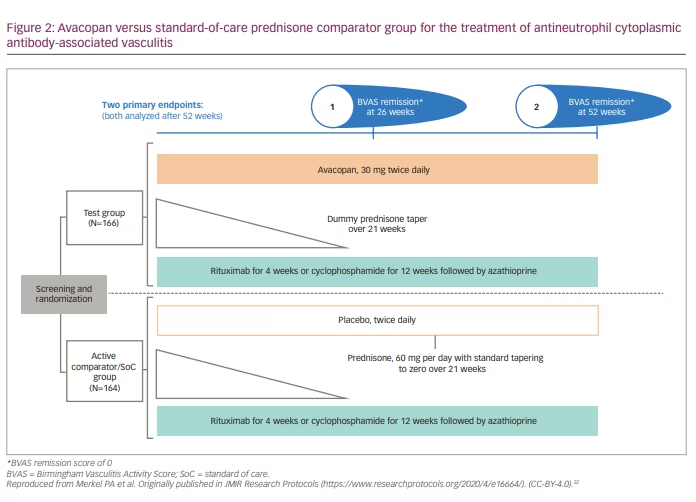
Patients received either avacopan 30 mg twice daily or placebo for 52 weeks. Prednisone or dummy prednisone were tapered over 21 weeks (Figure 2). For adults weighing more than 55 kg, the starting dose of prednisone was 60 mg daily; it was less for adults <55 kg, as well as for adolescents. The mean total prednisone-equivalent dose (oral + IV) was significantly lower in the avacopan group (1,349 mg total, equal to 4 mg/patient/day) compared with the prednisone group (3,655 mg, equal to 12 mg/patient/day). None of the patients received maintenance rituximab.
Endpoints
Primary endpoints were (1) clinical remission of AAV at week 26, as defined by a BVAS score of 0 and no receipt of glucocorticoids for 4 weeks before week 26, and (2) sustained remission of AAV, defined as remission at weeks 26 and 52 and no receipt of glucocorticoids for 4 weeks before week 52.
Secondary endpoints included: (1) glucocorticoid-induced toxic effects; (2) BVAS of 0 at week 4; (3) change from baseline in health-related quality of life; (4) relapse; (5) change from baseline in eGFR; (6) urine albumin: creatinine ratio; (7) urinary monocyte chemoattractant protein 1: creatinine ratio; and (8) vasculitis damage index (range 0–64, with higher scores indicating more damage).
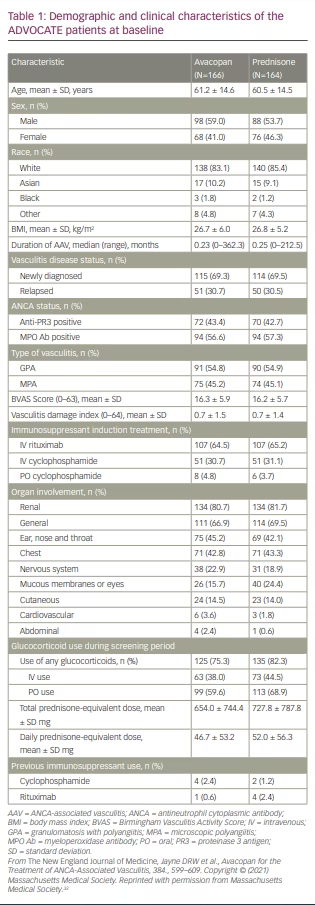
Demographic and clinical characteristics of patients at baseline
Patients had similar baseline characteristics (Table 1). The mean age was 60–61 years. Slightly more patients were male than female. The majority of patients identified themselves as white. About 69% in both groups had newly diagnosed disease and ~30% had relapsed disease. More patients in both groups had GPA (~54%) compared with MPA (~45%), despite greater prevalence of the MPO antibody (56.6% and 57.3% for avacopan and prednisone groups, respectively) than PR3 antibody (43.4% and 42.7% for avacopan and prednisone groups, respectively). BVAS and vasculitis damage index scores were similar between the two groups; the baseline mean BVAS was ~16 for both groups. The majority of patients received rituximab as induction therapy (64.5% and 65.2% for the avacopan and prednisone groups, respectively) compared with cyclophosphamide (total combined IV or oral cyclophosphamide 35.5% and 34.8% for the avacopan and prednisone groups, respectively). Organ involvement was similar in both groups; ~81% had evidence of renal involvement and ~43% had lung involvement. Glucocorticoid use during the screening period was similar between groups.
Results
The primary endpoints were disease remission defined as a BVAS of 0 and no glucocorticoid use in the previous 4 weeks at weeks 26 and 52. The first primary endpoint (remission at week 26) was observed in 120 of 166 patients (72.3%) receiving avacopan and in 115 of 164 patients (70.1%) receiving prednisone, which was not statistically significant in meeting a prespecified definition of noninferiority (estimated common difference, 3.4 percentage points; 95% confidence interval [CI], –6.0 to 12.8; p<0.001 for noninferiority; p=0.24 for superiority). Sustained remission at week 52 was observed in 109 of 166 patients (65.7%) receiving avacopan and in 90 of 164 patients (54.9%) receiving prednisone, which was statistically significant (estimated common difference, 12.2 percentage points; 95% CI, 2.6 to 22.3; p<0.001 for noninferiority; p=0.007 for superiority).
One of the secondary endpoints assessed glucocorticoid toxicity using the Glucocorticoid Toxicity Index (GTI), with higher scores representing greater toxicity. This novel index measures a number of glucocorticoid-related side effects, such as changes in body weight, blood pressure, myopathy, glucose tolerance and infections. Patients in the avacopan group had statistically lower GTI scores at both 13 weeks and 26 weeks compared with the prednisone group.
The ADVOCATE trial also showed a greater improvement in renal function glomerular filtration rate in the avacopan group compared with the prednisone group. At 1 year, 16 of 158 patients (10.1%) in the avacopan group had a relapse of their AAV compared with 33 of 157 patients (21.0%) in the prednisone group, which was statistically significant (hazard ratio for relapse, 0.46; 95% CI, 0.25 to 0.84).
Health-related quality of life, another secondary endpoint, was assessed with two validated standardized scoring systems: the 36-Item Short Form Health Survey (SF-36) and the EuroQoL Group 5-Dimensions 5-Level Questionnaire (EQ-5D-5L). Results favoured the avacopan group over the prednisone group.
Regarding safety, there were 116 serious adverse events in the avacopan group compared with 166 in the prednisone group. There were more deaths (4 patients [2.4%] versus 2 patients [1.2%]), life-threatening or serious adverse events, overall infections (124 [75.6%] versus 113 [68.1%]) and serious opportunistic infections (11 [6.7%] versus 6 [3.6%]) in the prednisone group compared with the avacopan group. Importantly, there were no cases reported (in either group) of Neisseria meningitidis infection. Slightly more patients in the avacopan group (9 patients [5.4%]) had abnormal liver function tests compared with patients in the prednisone group (6 patients [3.7%]); however, these resolved upon withdrawal of avacopan. Another recent smaller study also confirmed the safety of avacopan in 42 patients with AAV.35
Study conclusions
The current standard of treatment for patients with newly diagnosed AAV includes either cyclophosphamide or rituximab with tapering doses of glucocorticoids, followed by a maintenance dose of rituximab at 6 months. High doses of glucocorticoids are often used for remission induction. Avacopan, an oral C5a receptor antagonist, appears to be an effective glucocorticoid-sparing adjunctive treatment in conjunction with rituximab or cyclophosphamide used for the induction treatment of severe AAV. Avacopan was superior to a 26-week tapering schedule of prednisone with respect to remission of AAV at 52 weeks. Furthermore, patients in the avacopan group had fewer glucocorticoid-associated adverse events compared with the prednisone group and, despite complement inhibition, there were no cases of Neisseria meningitidis infection reported in either group. A caveat to this study is that all participants received glucocorticoids initially, albeit the total dose was much lower in the avacopan group. Future investigations might consider whether avacopan could supplant the use of glucocorticoids altogether for the induction treatment of AAV, and whether there might also be a role for avacopan in the management of minor disease relapses. It is also important to establish whether avacopan may add any additional benefits to currently accepted maintenance regimen with lower doses of rituximab.
US Food and Drug Administration concerns
On 6 May 2021, an FDA advisory committee narrowly voted in favour of supporting approval of avacopan for adult patients with AAV (10:8 in favour of approval).36 However, when the question of whether efficacy data were supportive of approval was raised, the committee remained split 9:9. While the ADVOCATE trial should be considered as a milestone in the treatment of AAV (GPA and MPA),37 demonstrating clear evidence of efficacy of avacopan (including decreased numbers of relapses, quality of life benefits and improved glomerular filtration rates) in addition to diminished glucocorticoid-related toxicities, the reviewers’ concern was that the study was aimed at demonstrating the efficacy of avacopan in both remission induction and remission maintenance using a study design where patients in remission were not assigned to a standard maintenance therapy with rituximab. This may have been the intentional choice of the drug manufacturer to compare the efficacy of avacopan over placebo in the second part of the study when the glucocorticoid dose was completely stopped in the glucocorticoid limb of the study (weeks 26–52). One of the important remaining questions is whether the risk of relapse in patients taking avacopan is different from the risk of relapse in patients on maintenance rituximab. Concerns were also raised about possible hepatotoxicity and superiority data at week 52 when non adjudicated data were analysed statistically. On 8 October 2021, the FDA approved avacopan (Tavneos®; ChemoCentryx, Inc.) as an adjunctive treatment to standard therapy for adults with severe active AAV. Avacopan is expected to have a wholesale price of $150,000–200,000 per patient per year.38