Diffuse alveolar haemorrhage (DAH) is associated with high morbidity and mortality. The causes of DAH are diverse, and include rheumatologic conditions such as granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), Systemic lupus erythematosus (SLE) and Goodpasture’s syndrome (Table 1). Rheumatologists are frequently consulted, given the high degree of suspicion for these underlying aetiologies. Another possible but rarely considered cause of DAH is Lane–Hamilton syndrome (LHS). LHS is the coexistence of coeliac disease (CD) with idiopathic pulmonary hemosiderosis (IPH). In this article, we describe a case of LHS presenting with DAH, in which anti-neutrophilic cytoplasmic antibodies (ANCA) testing was positive for atypical perinuclear ANCA (p-ANCA). LHS is an important vasculitis mimic that rheumatologists should be aware of when evaluating a patient with DAH.
Table 1: Causes of diffuse alveolar haemorrhage
|
DAH with capillaritis |
DAH without capillaritis (bland haemorrhage) |
|
Primary immune-complex vasculitis |
Idiopathic pulmonary hemosiderosis |
|
Goodpasture’s syndrome |
Antiphospholipid antibody syndrome |
|
Henoch-Schoenlein purpura |
Coagulopathy |
|
Primary pauci-immune small vessel vasculitis |
Pulmonary venous hypertension |
|
GPA |
Arterio-venous malformation |
|
MPA |
Pulmonary embolism |
|
EGPA |
Pulmonary veno-occlusive disease |
|
Isolated pulmonary capillaritis |
Mitral stenosis |
|
Secondary immune-complex vasculitis |
Acute respiratory distress syndrome |
|
SLE |
Sepsis |
|
Mixed connective tissue disease |
Bone marrow/lung transplantation |
|
Rheumatoid arthritis |
Cytotoxic drug therapy |
|
Diffuse connective tissue disease |
Radiation therapy |
|
Cryoglobulinemic vasculitis |
|
|
Drug induced (i.e. hydralazine, propylthiouracil) |
|
DAH = diffuse alveolar haemorrhage;EGPA = eosinophilic granulomatosis with polyangiitis;GPA = granulomatosis with polyangiitis;MPA = microscopic polyangiitis;SLE = systemic lupus erythematosus.
Case report
Our patient, a 54-year-old man, initially presented to the emergency room, with a history of 1 week of worsening hemoptysis. He also reported several months of progressive exertional dyspnoea, fatigue and generalized weakness. He denied fever, skin rash, sinus pain, nasal crusting, hearing impairment, neurologic symptoms, history of melena, easy bruising or bleeding from any orifice. He further denied any joint pains, lymph node swellings, weight loss or night sweats. He was a nonsmoker and denied a history of recurrent chest infections, recent travel or hospital admissions. There was no history suggestive of photosensitivity, alopaecia, oral ulcers, Raynaud’s phenomenon or dysuria. Prior to his presentation to the emergency room, he was diagnosed with iron-deficiency anaemia and treated with blood transfusions.
At admission, the patient was alert, and haemodynamically stable, with blood pressure 142/77 mmHg, low-grade fever with T max 99.9o °F (37.72 °C) and tachycardia with heart rate of 112 bpm. Skin pallor was noticed. He quickly deteriorated with worsening respiratory failure. Oxygen saturation dropped to 60%, on room air, therefore the patient required intubation on day 1 of admission. On examination, he had coarse bilateral crackles on auscultation and the heart rhythm was regular, without murmurs. The abdominal exam was benign overall, with no evidence of hepatosplenomegaly. He had no skin rash, no evidence of joint inflammation and no apparent sinus disease.
Investigations (presented in Table 2) showed an initial haemoglobin value of 12.1 g/dL, followed by an acute drop to 7.8 g/dL. The red blood cells in peripheral smear were microcytic and hypochromic with aniso/poikilocytosis suggestive of iron-deficiency anaemia. His serum iron (20 μg/dL) and iron saturation (6%) were low, while the total iron-binding capacity was normal (383 μg/dL). His platelets were elevated (457,000/mm3). Ferritin was also elevated (829.6 μg/L) in the setting of acute inflammation. His liver and renal function tests, oeosinophil counts, urine analysis and coagulation studies were all normal. Sputum examination for acid-fast bacilli was negative.
Table 2: Laboratory findings in our patient with Lane–Hamilton syndrome
|
Laboratory test |
Result |
Normal range |
|
WBC count |
10.7 k/mm3 |
3.7–10.5 k/mm3 |
|
RBC count |
3.08 m/mm3 |
4.50–6.20 m/mm3 |
|
Haemoglobin |
7.8 g/dL |
13.2–17.7 g/dL |
|
Haematocrit |
27% |
40–52% |
|
Mean corpuscular volume |
78 fL |
82–89 fL |
|
Mean corpuscular haemoglobin |
24 pg |
25–35 pg |
|
Platelet count |
457 k/mm3 |
150–400 k/mm3 |
|
Creatinine |
0.9 mg/dL |
0.6–1.2 mg/dL |
|
Erythrocyte sedimentation rate |
97 mm/hr |
0–15 mm/hr |
|
C-reactive protein |
14.4 mg/dL |
<0.5 mg/dL |
|
Ferritin |
829.6 ng/mL |
30.0–400.0 ng/mL |
|
Iron |
20 ug/dL |
59–158 ug/dL |
|
Iron saturation |
6% |
20–50% |
|
Total iron-binding capacity |
383 ug/dL |
235–451 ug/dL |
|
Thyroid stimulating hormone |
2.24 mcIU/mL |
0.27–4.20 mcIU/mL |
|
HIV |
<20 IU/mL |
<20 IU/mL |
|
Rheumatoid factor |
<10 IU/mL |
<14 IU/mL |
|
ANA |
<1:80 |
<1:80 |
|
Atypical p-ANCA |
Strong positive |
Negative |
|
Anti-MPO |
0.4 AI |
0.0–0.3 AI |
|
Anti-PR3 |
<0.2 AI |
0.0–0.3 AI |
|
Tissue transglutaminase IgA |
240.2 U/mL |
0.0–15.0 U/mL |
|
Anti-GBM |
<0.2 AI |
0.0–0.3 AI |
|
Anticardiolipin IgG/IgM |
<1.6; 4.4 U/mL |
<20.0 U/mL |
|
Anti-beta2-glycoprotein-1 IgG/IgM |
<1.4; 1.0 U/mL |
<20.0 U/mL |
|
Lupus anticoagulant |
Negative |
Negative |
ANA = antinuclear antibodies;GBM = glomerular basement membrane;IgA = immunoglobulin A;IgG = immunoglobulin G;IgM = immunolglobulin M;MPO = myeloperoxidase;p-ANCA = perinuclear anti-neutrophil cytoplasmic antibody;PR3 = proteinase 3;RBC = red blood cell;WBC = white blood cell.
Chest X-ray showed bilateral interstitial infiltrates (Figure 1a). Contrast-enhanced computed tomography showed extensive bilateral ground glass opacities and consolidative changes (Figure 1b). Bronchoscopy showed significant inflammation of the mucosa and friable airways, with progressive bloody fluid return on serial broncho-alveolar lavage (BAL), thus confirming the suspicion of DAH. BAL further revealed hemosiderin-laden macrophages (Figure 2) and, therefore, the patient was diagnosed with IPH.
Figure 1: Chest images
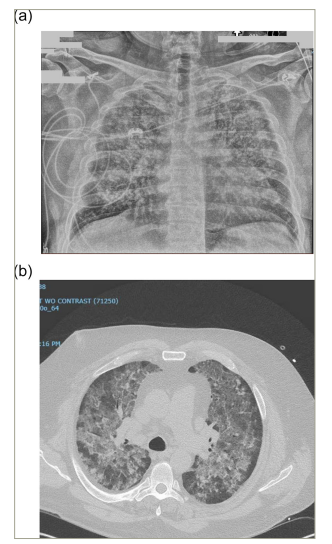
Chest anteroposterior view showing bilateral interstitial infiltrates (a); and computed tomography scan of the chest showing diffuse bilateral ground glass opacification of the lungs with interstitial thickening suggestive of diffuse alveolar haemorrhage (b).
Figure 2: Broncho-alveolar lavage sample with a Prussian blue-stained section that confirms hemosiderin (blue staining) in the histiocytes (alveolar macrophages)
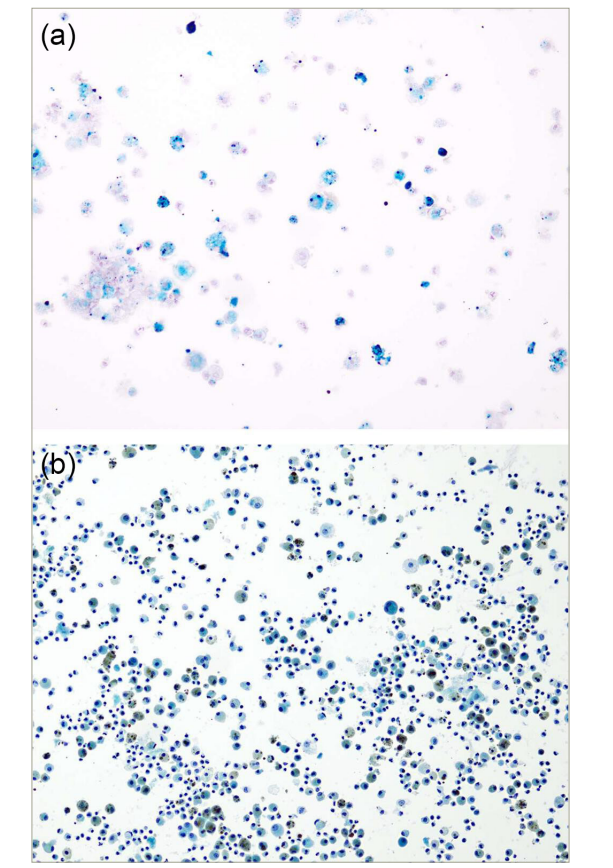
Broncho-alveolar lavage sample with a Prussian blue-stained section that confirms hemosiderin (blue staining) in the histiocytes (alveolar macrophages) i.e. hemosiderin ladenmacrophages (HLM) (a). Broncho-alveolar lavage sample with a Papanicolaou stain, which shows histiocytes (alveolarmacrophages) filled with brown granular material i.e. HLM (b).
Serum markers of auto-immune disorders including rheumatoid factor, anti-nuclear antibodies (ANA), anti-cardiolipin antibody, cryoglobulins and anti-glomerular basement membrane antibodies all returned negative. However, ANCA were positive at 1:320 titer with an atypical p-ANCA pattern. Myeloperoxidase antibody was borderline positive (0.4; N=0.0–0.3), while the proteinase 3 antibody was negative.
The patient’s DAH was managed with mechanical ventilation and sedation and systemic pulse corticosteroids. For the acute drop in haemoglobin complicating his acute respiratory failure, the patient required transfusion with 4 units of packed red blood cells.
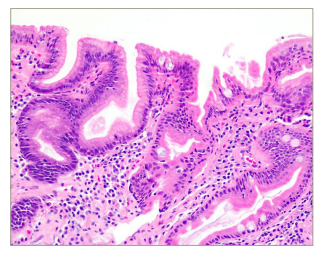
No cause was known for the prior finding of iron-deficiency anaemia in the patient. CD testing revealed positive serum anti-tissue transglutaminase antibody (anti-tTG immunoglubulin A [IgA]) (240.2 IU/ml, normal <15 IU/ml). Duodenal biopsy showed chronic active duodenitis (Marsh grade I) consistent with CD (Figure 3). Over his hospital course he was extubated and eventually weaned to room air. He was recommended to follow a gluten-free diet and received a course of tapering prednisone. Four months after discharge his haemoglobin was 13.2 g/dL.
Figure 3: Duodenal biopsy with haematoxylin and eosin stained section showing foveolar metaplasia, neutrophils and lymphocyte aggregates consistent with chronic active duodenitis
Over the next 6 years, however, he had relapses of severe dyspnoea, with imaging findings of DAH, sometimes with frank hemoptysis. In addition to repeated courses of high-dose prednisone and chronic low-dose prednisone, he was started on azathioprine.
Anti-tTG antibodies remained elevated 4 years after his initial hospitalization for DAH. His last episode of DAH occurred at 5 years. At 6 years after his initial diagnosis, he was hospitalized for acute respiratory failure requiring ventilatory support but with stable haemoglobin and no imaging evidence of DAH at that time. Anti-tTG antibodies were negative. One year later (7.5 years from diagnosis) he was off prednisone, and is now maintained on mycophenolate mofetil.
Discussion
Patients with DAH are typically critically ill. Systemic features such as fevers and malaise can affect up to half of patients with DAH.1,2 DAH may be secondary to a multitude of underlying conditions, including pulmonary vasculitis secondary to MPA, GPA, Goodpasture’s syndrome or SLE, to name just a few. Table 1 shows a comprehensive list of DAH causes. Our patient presented with DAH and low-grade fever closely mimicking systemic vasculitis. Even more complicating was the fact that he tested positive for ANCA but had no other features of systemic vasculitis. Interestingly, he tested positive for ANCA, but in an atypical p-ANCA pattern, not a classic cytoplasmic ANCA or p-ANCA pattern more commonly associated with systemic ANCA-associated vasculitis (AAV).
The condition of recurrent pulmonary haemorrhage that cannot be assigned to an underlying condition is usually defined as IPH. IPH is described as a triad of recurrent hemoptysis, iron-deficiency anaemia and infiltrates on chest radiographs. The association between IPH and CD was first observed by Lane and Hamilton in 1971 when authors described the clinical course of a 23-year-old man who presented with recurrent iron-deficiency anemia and failure to thrive since age 9.3 Barium studies performed at age 9 showed evidence of duodenal scarring. By the age 18, chest imaging studies showed persistent lung infiltrates. Lung biopsy was characteristic of IPH.3 The combination of these two conditions (IPH and CD), subsequently known as LHS, is extremely rare. To date, an estimated 80 cases have been reported.4 CD is an autoimmune enteropathy triggered by gluten ingestion in genetically predisposed individuals.5 Patients with CD, if symptomatic at all, have a highly variable clinical spectrum ranging from typical gastrointestinal manifestations, such as abdominal bloating, chronic diarrhoea, or steatorrhoea, to extraintestinal manifestations such as osteopenia, ataxia, liver abnormalities, infertility, dermatitis herpetiformis, etc.5 Although the prevalence of CD has increased over the past five decades, with a current estimate of 1% of the population worldwide,6 the prevalence of IPH among patients with CD remains unknown. CD should always be considered in the differential diagnosis of pulmonary hemorrhage, but general awareness of this condition is lacking.7
Exacerbations of IPH in LHS can lead to severe acute clinical deterioration, with hypoxaemia requiring ventilatory support and blood transfusion. Multiple recurrences can lead to restrictive lung disease, pulmonary fibrosis, and significant morbidity and mortality. IPH causes hypoxic respiratory failure in the acute setting, which leads to restrictive lung disease due to pulmonary fibrosis over time.8 It is estimated that prognosis is generally poor without early intervention with a mortality rate as high as 14% due to IPH in the acute phase.4
Diagnosis of IPH begins with clinical presentation, imaging and laboratory tests supporting pulmonary haemorrhage, and is confirmed by characteristic histopathological findings. Laboratory testing should be done to rule out other causes of IPH, such as AAV, but should also include testing for anti-tTG to confirm or exclude CD as an underlying contributor. This is especially important if pulmonary haemorrhage is preceded by a history of iron deficiency, a common sign of chronic malabsorption of iron. Indeed, anaemia seems to be the most prevalent problem secondary to chronic malabsorption and alveolar haemorrhage in LHS.
Although LHS is a combination of IPH with CD, typical gastrointestinal symptoms are not commonly observed in most case reports.9–12 Fewer than 20% of cases were found to have significant gastrointestinal symptoms at the time of IPH diagnosis.2 Thus, one should not fully rely on the presence or absence of gastrointestinal clinical symptoms when approaching patients presenting with DAH.
Serologic assays for CD are readily available and are highly sensitive and specific.13 Testing for anti-tTG is the most commonly used screening test. Diagnosis of CD is confirmed by small bowel biopsy: the classical histopathologic findings are infiltration of the lamina propria with plasma cells, neutrophils and eosinophils, intraepithelial lymphocytosis, crypt hyperplasia and villous blunting.14
The most common presenting manifestation of LHS in adults is hemoptysis.2 Chest imaging may show diffuse bilateral ground-glass opacities with mid and lower lung zones more commonly affected than the upper zones.4 Lung biopsy remains a gold standard for the diagnosis but, according to various case reports, this has not been the universal practice.2,4 Transbronchial biopsies and cryo-biopsy were procedures most often performed instead of a surgical lung biopsy, which is considered to carry increased overall risk. The histopathology usually reveals blood-filled alveolar spaces, free and intracytoplasmic hemosiderin macrophages, alveolar thickening with hemosiderin deposition, type 2 pneumocyte hyperplasia and absence of necrosis, capillaritis, granulomatosis and immune complex deposition.15 A more common practice is use of BAL to identify hemosiderin-laden alveolar macrophages and to demonstrate progressive bloody fluid return on serial lavage. Prussian blue staining revealing 10% or more macrophages to be hemosiderin-laden is considered a positive finding (Figure 1).
Tryfon et al. recently evaluated 80 cases of LHS by searching an international database.4 Of the 80 patients, 44 patients were children and 36 were adults. The most predominant feature was hemoptysis with associated iron-deficiency anaemia. Diffuse ground-glass opacities were the most common chest imaging finding in adults.4
Like CD, IPH is believed to be an immune-mediated disease, yet the pathogenesis is still unknown. It has been suggested that the common link between the two conditions may stem from the deposition of circulating immune-complexes containing food allergens on the basal membrane of alveolar capillaries.16 Indeed, Saha et al. reported a 25% prevalence of CD among adults with IPH.2
In the evaluation of patients with DAH, ANCA testing is considered crucial as management and prognosis of AAV differs significantly from other causes of DAH. Autoantibodies, other than those specific to CD are not commonly reported in most cases of LHS.2 While ANCA positivity has been reported in cases of paediatric-onset LHS,7,17 this has not, to our knowledge, been reported in a previously published case of adult-onset LHS in the literature.
ANCA positivity is rarely reported in patients with IPH.18 Saha et al. found that among a small series of published cases of IPH in which autoantibodies were tested, out of 38 patients, only 5 had evidence of autoantibodies; and of the 37 tested for ANCA, only one was positive.18 Based on the paediatric French RespiRare® cohort multiple antibodies including ANCA were found to be positive (anti-smooth-muscle antibodies was 50%; ANA was 45%; and ANCA was 40% of the tested patients).7
There has been prior observation of atypical p-ANCA in patients with CD. Freeman et al. found atypical p-ANCA antibodies in 6 out of 40 patients with biopsy-confirmed CD.19 Similarly, Damoiseaux et al. found atypical p-ANCA in 8 of 37 or 22% of patients with CD.20 As LHS is a rarely reported complication of CD, there are no studies or reports to indicate if atypical p-ANCA is more or less frequently observed in this subset but is plausible to assume an atypical p-ANCA can be identified.
The finding of atypical p-ANCA, unlike classic p-ANCA and c-ANCA, is not commonly associated with vasculitis or vasculitic causes of DAH.21 The finding of atypical p-ANCA in an adult patient with new-onset alveolar haemorrhage should instead raise concern for underling CD.
LHS is a rare disease for which no standard treatment has been formulated. A gluten-free diet is necessary, but this is not always sufficient in long-term management.22,23 Among cases reported, systemic corticosteroids have been considered as a first-line therapy in acute phases. The patient reported by Lane and Hamilton was unwilling to adhere to a gluten-free diet and was treated with azathioprine.3
Subsequently, those patients that were unable to wean off steroids or were refractory to steroids have been treated with steroid-sparing agents, mainly with azathioprine.7,24 Ioachimescu et al. recommended that azathioprine in combination with corticosteroids might be the best therapeutic regimen, especially in preventing IPH exacerbations.25 Case reports have described relapses, even when patients were maintained on azathioprine and high-dose steroids, especially if patients do not adhere to a gluten-free diet.4,12
Reading et al. were the first to report improvement of pulmonary symptoms with gluten-free diet in IPH.26 Since then, it has observed that in the majority of patients with LHS syndrome, gluten-free diet may lead to eventual drug-free remission.22,23,27 Thus, correct diagnosis of the underlying cause of IPH is essential in improving survival given the approach in treating LHS differs from that for vasculitic or truly idiopathic cases. Immunosuppressive therapy with cyclophosphamide or rituximab as used to treat AAV is not currently used for LHS where treatment recommendations center around a gluten-free diet. Clinical awareness of LHS, early diagnosis of underlying CD and prompt institution of a gluten-free diet can dramatically change the course of this potentially lethal disease.
Conclusion
LHS syndrome is a rare clinical entity that remains underdiagnosed secondary to lack of physician awareness. LHS presents with DAH which can convincingly mimic vasculitis. Prompt diagnosis and institution of a gluten-free diet is critical to improve survival outcomes. With the availability of relatively simple screening test for anti-tTG antibodies, it is recommended to screen all patients with DAH for CD24,28 even in those patients lacking GI symptoms.2 Given the frequent finding of atypical p-ANCA antibody in patients with CD, an atypical p-ANCA presence should raise suspicion for LHS in those with alveolar haemorrhage rather than vasculitis as the underlying cause.