The seronegative spondyloarthritides are a group of inflammatory arthropathies that are related in aspects of their clinical and radiographic presentations and genetic predisposition. Psoriatic arthritis (PsA) and axial spondyloarthritis (AxSpA) are two of the major diseases classified under this umbrella, in addition to reactive arthritis, inflammatory bowel disease-associated arthritis and undifferentiated spondyloarthritis. PsA is an inflammatory arthritis that occurs in approximately one-third of patients with psoriasis.1 It is a heterogeneous disease, and can involve the peripheral and axial joints, entheses, skin, gastrointestinal tract and eyes. AxSpA refers to spondyloarthritides with predominantly inflammatory axial involvement that meet the Assessment of SpondyloArthritis International Society (ASAS) classification criteria.1 It is further classified into radiographic (r-AxSpA) and non-radiographic (nr-AxSpA) forms, with the former implying inflammatory disease involving the sacroiliac joints and/or spine evident on radiographic imaging.1
Although PsA and AxSpA have several overlapping clinical features, their treatment can greatly vary. At the present time, a multitude of approved medications are available for the treatment of both, but may not have the same effect on each patient or presentation. With these multifaceted diseases, targeted therapy offers opportunities in the ‘treat-to-target’ philosophy, with the goal of disease remission based on articular and extra-articular involvement. Non-pharmacological therapies, such as smoking cessation, weight loss, physical therapy and exercise, are typically used in conjunction with non-steroidal anti-inflammatory drugs (NSAIDs) in the initial and less severe stages of disease.1 The cornerstones of disease management for both PsA and AxSpA are immunosuppressive agents, such as non-biologic and biologic disease-modifying anti-rheumatic drugs (DMARDs), as outlined by the American College of Rheumatology (ACR) guidelines.2,3 The decision of which medication to use in a patient largely depends on the individual’s disease presentation, insurance approval and comorbid conditions. Conventional DMARDs (cDMARDs), such as methotrexate, sulfasalazine and leflunomide, are first-line therapies in the 2018 ACR guidelines for PsA . If disease activity persists, biologics are considered as the next step in therapy. Tumour necrosis factor (TNF) inhibition has been successful in reducing disease activity, with examples of anti-TNF agents including etanercept, adalimumab, certolizumab, golimumab and infliximab. Often, these drugs are used in combination with cDMARDs for an additive effect. In patients with predominantly axial disease activity in AxSpA, cDMARDs are found to be less useful and therefore biologics (i.e. anti-TNF therapies) are initiated after first-line conservative therapies such as NSAIDs have been exhausted.1,2
However, with the extensive heterogeneity of both diseases, not all medications target the same structures or have identical affects. Over the past 10 years, an increased understanding of the pathophysiology of PsA and AxSpA has been cultivated, leading to the development of novel biologic agents outside of anti-TNF therapies. Apremilast (a phosphodiesterase-4 inhibitor) and ustekinumab (an interleukin [IL]-12 and -23 inhibitor) have been developed, but do not seem to target all aspects of PsA.1 Since 2016, several more key proinflammatory cytokine-signalling pathways, such as T-cell activation and the IL-17, IL-12, IL-23, and Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway, have been used as targeted therapeutic agents in the spondyloarthritides.1 This comprehensive review discusses medications for PsA and AxSpA that have been approved by the US Food and Drug Administration (FDA) since 2016 (Figure 1), as well as those that are currently undergoing clinical trials.
T-cell co-stimulatory inhibition
Abatacept, a cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4)–immunoglobulin (Ig) human fusion protein, is a biologic that was approved for PsA in 2017. It works by binding to CD80 and CD86 on antigen-presenting cells, inhibiting CD28 co-stimulatory signalling in T cells and thereby preventing naïve T-cell activation and further inflammatory cytokine propagation.1,4
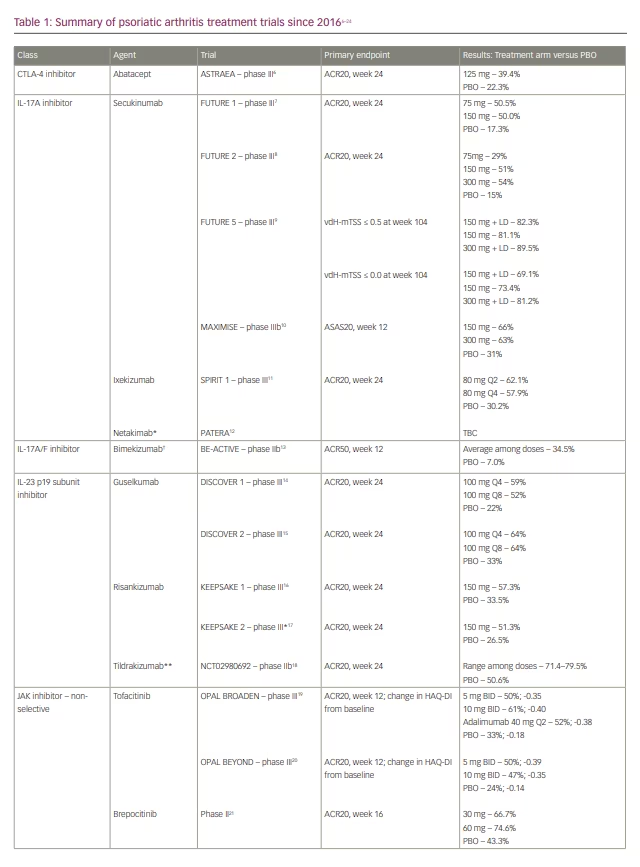
In the phase III ASTRAEA trial, patients who failed or were intolerant to at least one cDMARD with or without prior anti-TNF use were randomized to receive abatacept subcutaneous injections or placebo.5,6 The primary endpoint of a 20% improvement in the number of tender and number of swollen joints and a 20% improvement in three of five criteria (ACR20) at 24 weeks was seen in 39.4% of patients in the abatacept arm versus 22.3% in the placebo arm (Table 1).6-24 At week 16, those who did not achieve ACR20 responses were switched to open-label abatacept. After 24 weeks, all remaining patients were placed on open-label abatacept for an additional 28 weeks, and the ACR20 results became more equal between the two groups. Patients without previous anti-TNF use showed a greater response to abatacept than those with previous anti-TNF exposure (44% versus 36.4%). Other areas of improvement included enthesitis, dactylitis and, to a lesser degree, the minimal disease activity score and Composite Psoriatic Disease Activity Index. There was only a marginal difference in the proportion of patients with a 50% reduction in the Psoriasis Area and Severity Index (PASI 50) in the abatacept group versus placebo (26.7% versus 19.6%).5 Improvements in secondary endpoints, including improvement in physical function and lack of radiographic progression, were noted. Despite this, changes in Health Assessment Questionnaire–Disability Index (HAQ-DI) scores were deemed not statistically significant, and other secondary endpoints were therefore not further statistically analysed. Nail and axial disease were not evaluated.6
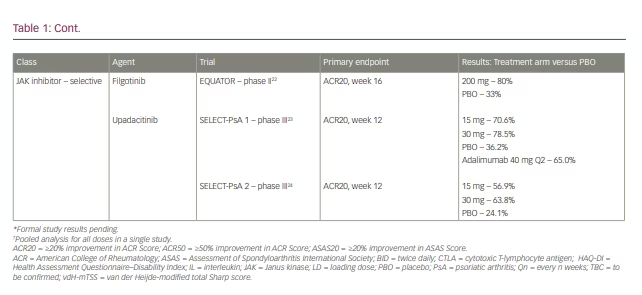
Despite its musculoskeletal benefits in PsA, abatacept is a fifth-line therapeutic agent in the 2018 ACR psoriatic arthritis guidelines.2 While there have been no head-to-head clinical trials, the more novel medications seem to elicit higher disease remission rates in more domains. 5 Furthermore, abatacept is a treatment that is often used in patients who need biologic therapy but are at high risk of infection, particularly in the absence of severe psoriatic disease.2,3 The notion of abatacept having a lower infectious risk is based on fewer hospitalizations secondary to infection in the rheumatoid arthritis population, in comparison to those on anti-TNF therapy.2,25 Pneumonia, bronchitis and upper respiratory tract infections are the most common adverse events, with a higher incidence in patients with chronic obstructive pulmonary disease.5 These data have been extrapolated to the PsA cohort.2 Abatacept poses an increased risk of chronic obstructive pulmonary disease exacerbations and must be used with caution in this population. It has not been shown to be teratogenic or carcinogenic.5
Abatacept has not been shown to be efficacious in AxSpA. In a small open-label pilot study conducted in 2011 in Germany, abatacept intravenous infusions were administered at 10 mg/kg on days 1, 15 and 29 and every 28 days thereafter to 15 anti-TNF-naïve patients (group one) and 15 anti-TNF inadequate responders (group two), with a primary endpoint of 40% improvement according to the ASAS criteria (ASAS40) at week 24.26 Although abatacept was well tolerated, only 27% of group one and none of group two reached the primary endpoint, with no change in C-reactive protein (CRP) levels, Patient Global Assessment scores or Bath Ankylosing Spondylitis Functional Index scores. Further analysis showed that 27% of group one and 20% of group two reached ASAS20, but the results were not significant enough to continue with further trials.26 To the best of our knowledge, no further major studies of abatacept in AxSpA have been conducted.
IL-17 cytokine axis
IL-17, a cytokine produced by T-helper 17 cells, is a well-known contributor to the pathogenesis of the spondyloarthritides.1 This specific cytokine family comprises multiple subproteins (A–F), with IL-17A and IL-17F being the most studied. Medications for the spondyloarthritides targeting IL-17 include blockade of these subproteins as well as the IL-17 receptor.27 Only IL-17A inhibitors are recommended for use in the current ACR treatment guidelines for PsA and AxSpA, particularly after failure of at least one anti-TNF medication.2,3 Other IL-17 cytokine axis inhibitors are not outlined in the guidelines at this time due to pending FDA approvals.2
IL-17A inhibitors
Secukinumab is an IgG1-kappa monoclonal antibody that was approved for the treatment of PsA in 2016.27 Its benefits for skin, peripheral and axial joint manifestations have been noted in the initial FUTURE trials and beyond. The FUTURE 1 trial showed the effectiveness of secukinumab for clinical symptoms and inhibition of structural damage from ongoing inflammation in patients with PsA. These patients all received an intravenous loading dose of secukinumab at 10 mg/kg at weeks 0, 2 and 4, followed by either 75 mg or 150 mg subcutaneous injections every 4 weeks against placebo starting on week 8 until week 24. Placebo patients were assigned to either secukinumab dosing at week 16 or 24 based on their clinical response.7 Initial trial results and a 5-year follow-up study showed sustained improvement in both skin and musculoskeletal domains.7,28 In FUTURE 2, secukinumab was evaluated at doses of 75, 150 and 300 mg every 4 weeks. Long-term extension data published in 2020 suggested sustained clinical outcomes spanning 5 years after completion of the FUTURE 2 trial (Table 1).8,29 The phase III FUTURE 5 trial was a 2-year study which demonstrated that secukinumab could potentially delay radiographic progression of PsA peripheral joint disease at both 300 mg and 150 mg dosages.9 Patients were initially divided into four groups: 300 mg with loading dose, 150 mg with loading dose, 150 mg without loading dose, and placebo. The first part of the study was to assess clinical efficacy of secukinumab, with results paralleling prior FUTURE studies. The second aspect was to evaluate radiographic changes. In this part, placebo patients were changed to either secukinumab 300 mg or 150 mg at week 16 (clinical non-responders) or week 24 (remaining patients) until the completion of the study. Radiographic progression was based on the mean change from baseline in van der Heijde-modified total Sharp score (vdH-mTSS) in radiographs of the hands, wrists and feet. A mean change from baseline vdH-mTSS of ≤0.5 or ≤0.0 would indicate no structural progression. Radiographs were obtained at baseline and again at week 16 (for clinical non-responders), 24, 52, and 104. The end results showed over 80% of patients in each group reached ≤0.5 and majority obtaining ≤0.0, signifying the benefit of seckinumab on a structural level (Table 1).9 Secukinumab was also found to be effective for PsA with axial manifestations in the MAXIMISE trial, with ASAS20 responses in up to 66% of participants treated with secukinumab versus 31% of those in the placebo arm at week 12 and a continued effect until week 52.10
To determine the efficacy of secukinumab against anti-TNF inhibitors, a head-to-head trial was conducted against adalimumab (the EXCEED trial).30 Participants in one arm of the trial received secukinumab 300 mg as a loading dose (at baseline, then at weeks 1, 2, 3 and 4) and every 4 weeks thereafter for 48 weeks, while those in the other arm received citrate-free adalimumab every 2 weeks for 40 weeks. At week 52, a similar proportion of patients in both study arms had reached the primary endpoint of ACR20: 67% for secukinumab and 62% for adalimumab (Table 2).30,31 Superiority of secukinumab to adalimumab was not statistically significant, although secukinumab did have a higher intention rate in disease control. The safety profiles of the two drugs were also similar and there were no new safety signals.30
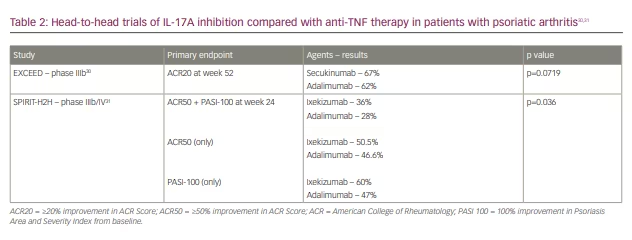
Ixekizumab, an IgG4-kappa monoclonal antibody, is an IL-17A inhibitor that has been FDA approved for use in PsA since 2017. The SPIRIT trial demonstrated a statistically significant improved ACR20 response with ixekizumab compared with placebo as early as week 2 in both anti-TNF-naïve and -exposed patients.11 Domains of improvement included disease activity, dactylitis and clearance of plaque psoriasis, while radiographic progression of axial spine disease was decreased. Improvements in enthesitis were also seen with ixekizumab, but were not statistically significant.11 An extension of this trial assessed ixekizumab head-to-head against adalimumab, with primary endpoints of ACR50 and PASI 100 at week 24.31 Other factors measured were arthritis, skin disease and quality of life. Overall, ixekizumab was non-inferior to adalimumab in ACR50 and superior in achieving PASI 100, with greater responses in arthritis, skin, nail disease and quality of life measures. Fewer serious adverse events were noted with ixekizumab (3.5%) versus adalimumab (8.5%) (Table 2).31
IL-17A inhibitors also appear to have significant effectiveness in AxSpA. Secukinumab was approved for the treatment of AxSpA after the MEASURE 1 and 2 trials (the former included an intravenous loading dose, while the latter was treated only with subcutaneous dosing), which demonstrated significant efficacy of secukinumab at 16 weeks and extending up to 5 years in post-marketing analyses.32,33 Ixekizumab use in AxSpA presented comparable results between patients who were biologic-naïve (COAST-V) and anti-TNF non-responders (COAST-W), leading to its approval for use in AxSpA in 2019 (Table 3).32-42 While COAST-V did include an adalimumab trial group, this was used mainly as an active reference group and not used for comparative analysis.34
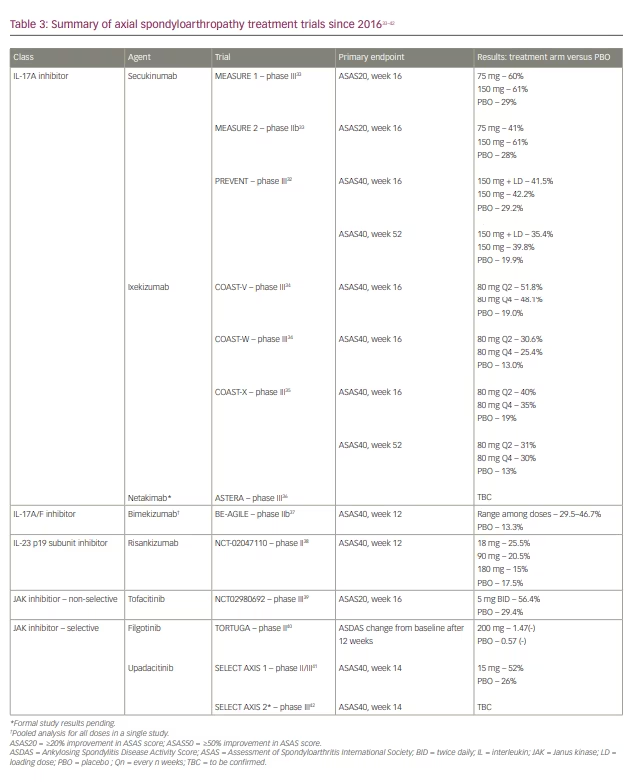
The aforementioned studies investigating IL-17A inhibitiors in AxSpA focused on radiographic progression and disease burden. More recently, trials exploring the benefits of IL-17A inhibitiors in nr-AxSpA disease have been conducted and published. The PREVENT trial included patients treated with secukinumab who were deemed as a failure or intolerant to a maximum of one anti-TNF, while the COAST-X trial investigated ixekizumab in patients who were biologic-naïve. Each focused on reduced signs and symptoms of disease burden, including inflammatory back pain, CRP levels, and resolution of sacroiliitis on magnetic resonance imaging. The data for both secukinumab and ixekizumab were significantly positive in comparison with placebo, leading to FDA approval in patients with nr-AxSpA in 2020 (Table 3).32,35
Both secukinumab and ixekizumab have been well tolerated in their clinical trials. Common side effects included respiratory infections and candidiasis. However, there is concern regarding inflammatory bowel disease flares and possible new-onset inflammatory bowel disease with their use. Reasons for this paradoxical effect have been hypothesized, but it ultimately requires further study.43
Netakimab is a third IL-17A humanized monoclonal antibody that is being investigated in a phase III clinical trial in PsA (PATERA). The trial has enrolled patients who have had an inadequate response to cDMARDs or one anti-TNF agent. The preliminary data published suggest that patients treated with netakimab have significant improvement in their total mobility, HAQ-DI score and overall pain compared with placebo arms. Completion of this study is projected to be in November 2022.12 A subanalysis of PATERA data thus far reported on the achievement of ACR20/50/70 in patients with PsA with and without inflammatory back pain (IBP) from weeks 8 to 24.44 Results were comparable in patients with or without IBP in achieving ACR20 at week 24 (81.5% versus 83.7%). However, fewer patients with IBP achieved both ACR50 and ACR70 compared with those without, indicating that netakimab may have limited benefit in axial disease.44 Its use in AxSpA is actively being studied in the phase III ASTERA trial, with preliminary sub-analysis data suggesting the utility of netakimab in symptomatic axial disease.36 The ASTERA trial is expected to be completed in 2022.45 Netakimab is currently approved for use in PsA and AxSpA in Russia and Belarus.29,36
IL-17A and IL-17F co-inhibition
Bimekizumab is an IgG1-kappa humanized monoclonal antibody that binds and neutralizes both the IL-17A and IL-17F subunits, thereby leading to a wider range of cytokine-signalling inhibition.27 In vivo, it has been found to be more potent than secukinumab and comparable to ixekizumab in inhibiting IL-17A. In phase I trials, ACR20/50/70 were maximal at weeks 8/12/16, respectively, in patients with PsA.13 The phase II BE-ACTIVE trial yielded improvement in skin, joints, enthesitis and HAQ-DI domains by week 48.13 The most common adverse events included viral upper respiratory infections and liver enzyme elevation.13
The phase III BE-COMPLETE trial, which aims to evaluate the safety and efficacy of bimekizumab in anti-TNF non-responders, is currently under way.13 While data for PsA are pending, a phase IIIb, head-to-head study of bimekizumab versus secukinumab in plaque psoriasis reported that bimekizumab was associated with greater positive changes in PASI scores over 16 and 42 weeks. It was ultimately deemed both non-inferior and superior to the IL-17A-specific inhibitor.46 However, the bimekizumab-treated group had higher events of mild-to-moderate oral candidiasis and neutropenia.46 FDA approval for the use of bimekizumab in psoriasis in the USA has been delayed due to the COVID-19 pandemic.47
BE-AGILE is an ongoing phase IIb study of various bimekizumab doses (16 mg, 64 mg, 160 mg and 320 mg every 4 weeks) in AxSpA that has shown promising preliminary data. Compared with placebo, higher proportions of patients reached the primary endpoint of ASAS40 at week 12 with all bimekizumab doses. At week 48, higher percentages of ASAS40 responses were seen in participants treated with bimekizumab 160 and 320 mg, compared with those treated with 16 mg and 64 mg.37
Anti-IL-23 p19 subunit
IL-23 is another integral player in the pathogenesis of the spondyloarthritides. IL-23 is a heterodimeric cytokine constituted of p19 and p40 subunits. Historically, only its p40 subunit was targeted in conjunction with IL-12 inhibition, such as in ustekinumab.48 More recently, monoclonal antibodies targeting the p19 subunit have shown efficacy in downstream signalling blockade, with convincing results in PsA management.
Guselkumab is an anti-IL-23 p19 monoclonal antibody that is approved for use in psoriasis and PsA in the USA. The VOYAGE 1 and 2 trials evaluated guselkumab in psoriasis, and demonstrated significant improvements in PASI scores (PASI90 and PASI100) by week 16 when against placebo and adalimumab.27 In the ECLIPSE trial, guselkumab notably showed non-inferiority to secukinumab with fewer side effects in patients with moderate-to-severe psoriasis.49 The DISCOVER 1 and 2 trials aimed to study the use of guselkumab in PsA specifically.27 DISCOVER 1 included patients with active PsA and an inadequate response to biologic anti-TNF inhibitors. The primary endpoint of ACR20 at week 24 was reached by a higher proportion of patients in both guselkumab arms – dosed every 4 weeks (59%) and every 8 weeks (52%) – compared with the placebo arm (22%).14,50 DISCOVER 2 included biologic-naïve patients treated with guselkumab every 4 or 8 weeks versus placebo and reported a higher percentage of patients reaching ACR20 with guselkumab versus placebo, regardless of the dosing interval. In a 1-year follow-up study, patients in the guselkumab arms had sustained improvement in skin, joints, dactylitis, enthesitis and overall quality of life.14,48,50 No direct comparative studies between guselkumab and IL-17A inhibitors or anti-TNF therapy in PsA have been completed at this time.
Risankizumab, another monoclonal antibody targeting IL-23 p19, is approved for use in plaque psoriasis in the USA and most recently for PsA.27,51 Pre-publication data presented at the European Alliance of Associations for Rheumatology and ACR conferences in 2021 suggested that patients treated with risankizumab met the primary endpoint of ACR20 at week 24 for the treatment of PsA in both the KEEPSAKE 1 trial (patients who had failed cDMARDs, 57.3% versus 33.5% in placebo arm) and the KEEPSAKE 2 trial (anti-TNF inadequate responders, 51.3% versus in 26.5% placebo arm).16,17 The dosage studied was 150 mg at weeks 0, 4 and 16 (Table 1). Secondary endpoints were also met, demonstrating improvement in skin, nail psoriasis, dactylitis, enthesitis and minimal disease activity scores. In early 2022, risankizumab was approved by the FDA for use in PsA.51 After loading doses at weeks 0 and 4, it is administered four times per year – the same regimen used in psoriasis.51
A third anti-IL-23 p19 monoclonal antibody, tildrakizumab, was studied in patients with PsA in a randomized phase IIb study with results published in 2021.18 Several doses of tildrakizumab at different intervals (200 mg every 4 weeks, 200/100/20 mg every 12 weeks) were compared with placebo (every 4 weeks), with a primary endpoint of ACR20 response at week 24. Beyond week 24, patients on placebo and tildrakizumab 20 mg every 12 weeks were switched to tildrakizumab 200 mg every 12 weeks until week 52. At week 24, a pooled analysis of the treatment arms showed an ACR20 response rate of 71.4–79.5% with tildrakizumab compared with 50.6% with placebo. The tildrakizumab arms also had higher rates of ACR50, Disease Activity Score 28-CRP and PASI scores beyond week 24. While improvements in joint and skin disease were seen, there was not as much benefit in dactylitis or enthesitis.18
Mirikizumab is the fourth member of this class and has been studied in the treatment of moderate-to-severe plaque psoriasis only. A phase II study published in 2019 found that PASI90 at 16 weeks was seen in 0% of patients given placebo, compared with 67%, 59% and 29% of patients receiving mirikizumab 300, 100 or 30 mg, respectively, every 8 weeks.52 This medication has yet to be studied in PsA.
Despite evidence that IL-23 acts upstream of T-helper 17 cells, as a whole, IL-23 p19 inhibitors have not been shown to provide clinical efficacy in AxSpA at the time of this review. Notably, risankizumab has been studied in a phase II clinical trial in AxSpA and, while CRP levels were reduced, ASAS40 at week 12 was not met in patients with active disease (Table 3).38 Various hypotheses have been put forward for this, with one suggesting that IL-23 cytokine signalling may not be involved in axial spine inflammation and axial enthesitis, and therefore its blockade did not yield notable effects.48,53 The lack of clinically efficacious gains requires further consideration.
Adverse events of the anti-IL-23 p19 class have been noted to be similar to those of placebo, with similar, low rates of serious infections.14,17 In phase II trials with guselkumab, a 5% chance of neutropenia has been noted.27 Basal cell carcinoma and acute myocardial infection have been reported with the use of risankizumab, but further studies are needed.27
JAK–STAT inhibition
JAKs are multidomain tyrosine kinases consisting of four subtypes: JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2).54 When cytokines bind to JAK receptors, JAKs phosphorylate themselves, their receptors and subsequently STATs. This constitutes the JAK–STAT pathway and activates downstream intracellular cytokine pathways. These signals are also known to stimulate other proinflammatory cytokines, such as IL-17 and IL-23, potentially inducing a much larger inflammatory response. Older JAK inhibitors target more than one subtype of JAK (‘non-selective’), while newer-generation JAKs are termed ‘selective’ and target only a single subtype.
Non-selective JAK inhibitors
In 2017, tofacitinib became the first JAK pathway inhibitor to be approved for use in PsA.27 It is non-selective, with the ability to inhibit both the JAK1 and JAK3 subtypes.54 Two phase III trials, OPAL BROADEN (anti-TNF-naïve patients) and OPAL BEYOND (anti-TNF non-responders), studied the effects of tofacitinib in PsA (Table 1). Primary endpoints were ACR20 and change in HAQ-DI at 3 months from baseline.53,54
As part of the OPAL BROADEN study, researchers included adalimumab use as a reference test group. Patients were randomized 1:1:1:1 to receive tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, adalimumab 40 mg every 14 days or placebo (later switching to tofacitinib 10 mg at 3 months). ACR20 and change in HAQ-DI at 3 months were significant in both tofacitinib arms (50%, -0.35 in 5 mg tofacitinib group; 61%, -0.40 in 10 mg tofacitinib group) compared with placebo (33%, -0.18). Results compared with adalimumab (52%, -0.38) were numerically similar to tofacitinib arms; however, superiority and inferiority were not formally evaluated between the two.19
The OPAL BEYOND trial was randomized using a 2:2:1:1 ratio consisting of tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, placebo arm switched to 5 mg twice daily at 3 months, and an additional placebo arm switched to 10 mg twice daily at 3 months. At the initial 3-month evaluation, ACR20 and HAQ-DI responses were similar between tofacitinib groups (5 mg – 50%, -0.39; 47%, -0.35) compared with combined placebo arms (24%, -0.14). At 6 months, significant improvements in placebo-to-tofacitinib groups were noted (5 mg – 50% -0.48; 10 mg – 54%, -0.42), compared with original tofacitinib treatment arms (60%, -0.44; 49%, -0.39). Not only did this study suggest superiority of JAK inhibitors compared with placebo at 3 months, but also demonstrated their sustained and additive effects over time.20
In both studies, primary endpoints were met with improvements in skin, enthesitis and dactylitis seen as early as week 2.19,20 Currently, tofacitinib (5 mg twice daily) is the only JAK inhibitor mentioned in the ACR guidelines for PsA, with a low conditional recommendation to begin tofacitinib after IL-17A inhibitors unless contraindications are present or oral therapy is preferred.2
The efficacy of tofacitinib in active AxSpA has been proven in a phase III study, in which 56.4% of tofacitinib-treated patients achieved ASAS20 at 16 weeks compared with 29.4% in the placebo arm.39 However, the FDA approval of tofacitinib for AxSpA was delayed by indications of an increased risk of major adverse cardiovascular events (MACE), venous thromboembolism and malignancy with tofacitinib versus adalimumab in patients with rheumatoid arthritis in post-marketing data.55 These were seen at both lower (5 mg twice daily) and higher (10 mg twice daily) doses of tofacitinib, despite previous suggestions of a better safety profile with lower doses. Patients included in this post-marketing analysis included those over 50 years old with at least one risk factor for heart disease. Incidence rates were higher in patients treated with tofacitinib compared with those treated with anti-TNF therapy after 4 years for both malignancy (excluding nonmelanoma skin cancers) and MACE (4.2% versus 2.9%; 3.4% versus 2.5%, respectively). In subgroup analyses, patients over the age of 65 and those living in North America had higher incidence rates for both cancers and MACE.56 In December 2021, tofacitinib was approved by the FDA for AxSpA, with focus on these newly recognized adverse events. Tofacitinib is now recommended as a third-line treatment for AxSpA after the failure of anti-TNF and IL-17A inhibitors.2
Adverse reactions with tofacitinib in PsA and AxSpA trials have been concordant with those seen in rheumatoid arthritis trials, and include pneumonia, viral infections (particularly herpes zoster, occurring more commonly in the Asian population), venous thromboembolism, lipid profile elevations and liver transaminitis.39,54 Gastrointestinal perforations have been reported to occur more often in tofacitinib versus placebo groups, and are usually seen in patients with background corticosteroid and NSAID use; therefore, a history of diverticulitis is a contraindication for use. Though infrequent, it is important to note there were small risks of malignancy, namely non-melanoma skin cancer and lymphomas, most predominantly non-Hodgkin’s B-cell lymphomas.19,20,57 The frequencies of such have yet to be defined.
At the 2021 ACR conference, brepocitinib, a combination JAK1–TYK2 inhibitor, was announced as another potential upcoming therapy for PsA. In a phase IIb trial comprising patients with an incomplete response to DMARDs, an ACR20 response was observed in 66.7% and 74.6% of participants given 30 or 60 mg brepocitinib, respectively, compared with 43.3% of placebo participants at week 16.21 Phase III trials of brepocitinib in both PsA and AxSpA are currently recruiting. This could add to the growing number of treatments within this class and lead to better control of disease activity.
Selective JAK and TYK inhibitors
Both filgotinib and upadacitinib are inhibitors of the JAK–STAT pathway and are selective in their inhibition of JAK1 only.27
Filgotinib was studied in a phase II trial (EQUATOR) in patients with moderate-to-severe PsA with an inadequate response to cDMARDs.22 Patients treated with filgotinib showed a significant improvement in the ACR20 response rate at week 16 versus placebo with changes noted as early as week 1, indicating a rapid onset of action and clinical benefit. Adverse reactions were similar to those in the placebo arm, and the number of viral infections was lower than that seen with tofacitinib. No malignancies or thromboembolic events were reported; however, the safety and efficacy of filgotinib need to be further studied in a phase III trial with longer duration.22
The benefit of upadacitinib in PsA was evident in the recently concluded phase III SELECT-PsA 1 (biologic-naïve patients) and SELECT-PsA 2 (biologic inadequate responders) trials.23,24 Both studies compared upadacitinib 15 mg and 30 mg daily against placebo, while SELECT-PsA 1 included an additional adalimumab control arm. The results of SELECT-PsA 1 showed both doses of the JAK inhibitor to be non-inferior to adalimumab in achieving ACR20 at week 12 (Table 1). Only upadacitinib 30 mg showed superiority to adalimumab, but at the cost of higher risks of serious adverse events (6.1%) compared with 15 mg (3.3%), placebo (3.1%) and adalimumab (3.7%).23 While there were improvements in enthesitis and inhibition of radiographic progression in both upadacitinib arms, the quantitative efficacy of dactylitis resolution was unable to be determined.24 In SELECT-PsA 2, statistically significant improvements were observed in both doses of upadacitinib compared with placebo in achieving primary and secondary endpoints, including ACR20 and ACR50/ACR70 at week 12, respectively (Table 1).24 Other domains showing benefit included PASI scores, HAQ-DI, physical function, and resolution of enthesitis and dactylitis.24
The most common adverse events reported in the SELECT-PsA studies were analogous to those seen with other JAK inhibitors. Pneumonia was the most severe adverse reaction, occurring in a single patient in the 15 mg group and six patients in the 30 mg group. There was a higher risk of opportunistic infections and herpes zoster in the higher dosage arm; however, risks of malignancies and lymphopenia were similar between dosages (three in each upadacitinib arm compared with zero in the placebo group).23,24 The FDA approval of upadacitinib was initially delayed while the risk of major cardiovascular events and cancers in patients with chronic inflammatory conditions were clarified; however, upadacitinib was ultimately approved for use in PsA in December 2021.55
The TORTUGA study was a phase II trial exploring filgotinib use in patients with active AxSpA who had a poor response to at least two oral NSAIDs.40,58 Patients on current cDMARD therapy were included, with the caveat that they were on therapy for at least 12 weeks and a stable dose for a minimum of 4 weeks. Patients who had previously been on anti-TNF therapy were required to complete a washout period before study initiation. The primary endpoint was change in Ankylosing Spondylitis Disease Activity Score (ASDAS) from baseline to week 12, either as a major or clinically significant change (ASDAS >2.0, ASDAS >1.1, respectively). 33% of patients treated with filgotinib experienced a major change in ASDAS compared with 2% treated with placebo. Furthermore, clinically significant ASDAS changes were noted in 66% of the study arm versus 40% in placebo. Overall, higher proportions of patients treated with filgotinib met primary endpoints compared with placebo, demonstrating its benefit in this inflammatory condition (Table 3).40
The SELECT-AXIS 1 trial was a phase II/III clinical study that evaluated upadacitinib 15 mg daily in patients with active AxSpA over 14 weeks.41 The primary endpoint of ASAS40 response was reached by 52% of patients in the upadacitinib arm compared with 26% in the placebo arm, with a rapid benefit and sustained efficacy for more than 2 years. No new safety signals were observed and the most common adverse reaction was asymptomatic elevation of creatinine phosphokinase. This trial did not evaluate patients with previous biologic use or those with nr-AxSpA.41 Further phase III trials are pending.58 In addition, the SELECT-AXIS 2 trial investigating upadacitinib efficacy in nr-AxSpA is now under way.42
Deucravacitinib is a recently developed selective TYK2 inhibitor, and patients with PsA and AxSpA are currently being recruited for trials. Data from a phase II study showed efficacy in meeting ACR20 responses compared with placebo at week 16.59 The primary adverse reactions included nasopharyngitis, sinusitis, headache and rash. No increased incidences of herpes zoster, thrombotic events or opportunistic infections were reported in the deucravacitinib group.59
Conclusion
Advances in therapeutic agents for the spondyloarthritides have led to a higher number of patients achieving minimal disease activity with low risk of adverse events. This article reviews the developments since 2016 in both PsA and AxSpA treatments, including current data providing evidence of efficacy. The decision of which medication to use largely depends on the patient’s insurance, individual patient and provider preferences, comorbid conditions, and the degree of articular and extra-articular disease involvement. While more therapies are being researched in clinical trials around the world, the advances made so far have been remarkable and will continue to improve the quality of life of these patients.