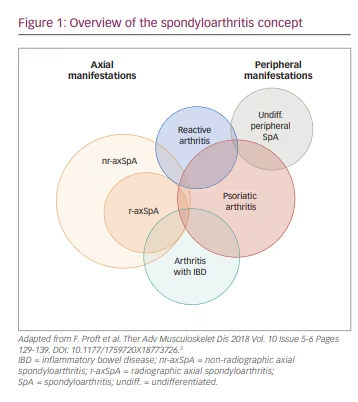
The term spondyloarthritis (SpA) encompasses a number of disease entities that share common clinical, biological and genetic characteristics. Common clinical features include inflammation in the axial skeleton, including the spine and sacroiliac joints, and inflammation of peripheral musculoskeletal structures such as arthritis, enthesitis and dactylitis.1 The disease is therefore divided by (predominantly) axial or peripheral manifestations. However, the disease can also be distinguished by more specific phenotypes, including axial spondyloarthritis (axSpA), psoriatic arthritis (PsA), arthritis with associated inflammatory bowel disease (IBD; i.e. Crohn’s disease and ulcerative colitis), reactive arthritis and undifferentiated SpA (Figure 1).2,3 These disease entities might also be associated with certain extra-musculoskeletal manifestations, including acute anterior uveitis, psoriasis or IBD. Furthermore, the concept of axSpA has undergone substantial modifications due to the developments in imaging, such as the use of magnetic resonance imaging for research and diagnostic purposes, which resulted in the implementation of the Assessment of SpondyloArthritis International Society classification criteria in 2009.4 Accordingly, axSpA is now sub-categorized into: radiographic axSpA (r-axSpA; formerly known as ankylosing spondylitis), characterized by the presence of radiographic changes in the sacroiliac joints and fulfilment of the modified New York criteria, and non-radiographic axSpA (nr-axSpA), which does not fulfil the modified New York criteria.3,5 According to the current understanding, the two entities of axSpA are considered a continuum of the same disease spectrum, ranging from nr-axSpA as an early disease stage identifier to r-axSpA as a later disease stage or more severe disease manifestation.3
The development of new treatment options in recent decades, such as biological disease-modifying anti-rheumatic drugs (DMARDs) and the resulting treatment responses for specific manifestations have made it evident that certain differences exist between the different disease entities of SpA. Some notable observations based on treatment efficacy were made, including that non-infectious uveitis and Crohn’s disease did not respond to anti-tumour necrosis factor (TNF) receptor blockade (e.g. etanercept),6,7 whereas interleukin (IL)-17A inhibition (e.g. secukinumab) showed a lack of efficacy in Crohn’s disease,8 and IL-23 inhibition (e.g. risankizumab) and IL-12/-23 inhibition (e.g. ustekinumab) only showed efficacy in PsA and not in axSpA.9,10 This growing understanding of the different disease entities of SpA has further supported the distinction of smaller disease entities.
Pathogenesis
Human leukocyte antigen B27
The pathogenesis of SpA remains unravelled despite significannt efforts. A strong association with a certain genetic predisposition – human leukocyte antigen (HLA)-B27 – was shown more than 50 years ago and is now well established.11,12 The association is most prominent for r-axSpA, as it accounts for 20–50% of the heritable component of the disease, whereas other genes explain approximately 4% of the heritability.12 Furthermore, the polymorphisms in certain other genes (e.g. ERAP1) are only associated with r-axSpA in HLA-B27-positive patients or patients carrying another risk allele for the disease (HLA-B40).13 HLA-B27 does not directly affect bone formation in transgenic mice in vivo or in human bone precursor cells in vitro, and the association with SpA might be caused by HLA-B27-driven inflammation.14 One hypothesis regarding HLA-B27 as a driver of inflammation involves the ability of HLA-B27 to present pathogen-derived arthritogenic peptides to CD8+ T cells, resulting in the selection of high-affinity autoreactive T cells.15,16 Another hypothesis is based on the presence of a free cysteine residue in HLA-B27 heavy chains, which promotes the formation of open conformations of HLA-B27, thereby affecting binding capacities for other immune receptors.17,18 Furthermore, the free cysteine residue, among other molecular properties of HLA-B27, promotes a tendency for HLA-B27 misfolding in the endoplasmic reticulum.19 Subsequently, disposal of the misfolded proteins can cause stress and promote the production of pro-inflammatory mediators and, hence, inflammation.20
Environmental trigger
Previous studies have identified the associations between preceding infections, such as gastroenteritis or chlamydia, and the development of reactive arthritis.21 The hypothesis of the involvement of the intestinal microbiome has been supported by the involvement of the intestines in disease manifestations, such as co-existing IBD as a part of the SpA disease manifestations.22 This was first described in the mid-1950s when Steinberg et al. published a series of case reports of co-existing SpA and IBD.23 Over the following years, it was widely acknowledged that a considerable overlap exists between SpA and IBD, especially Crohn’s disease. In Europe, the prevalence of IBD among patients with SpA is approximately 6–14%, which is elevated compared with an overall prevalence of IBD of 0.2%.24–26 Conversely, 20–30% of all patients with IBD develop arthritis resembling SpA, and this accounts for the most common extra-intestinal manifestation associated with the disease.27,28 Furthermore, up to 50% of all patients with SpA show microscopic signs of intestinal inflammation – most commonly without reporting any gastrointestinal symptoms.29 Most patients with SpA are treated regularly with non-steroidal anti-inflammatory drugs, which have been shown to increase the permeability of the intestines; the treatment has therefore been hypothesized to cause this phenomenon.30 The increased intestinal permeability has been observed in patients with rheumatoid arthritis (RA) treated with non-steroidal anti-inflammatory drugs and in patients with untreated SpA, thus the current evidence remains ambiguous.31,32 Specific microbial agents as triggers in the subtypes of SpA have also been investigated extensively throughout the years. However, the results of specific triggers have not yet been established.33 This aspect is further complicated by the fact that intrinsic predisposing factors, such as HLA-B27 positivity, might affect the microbiome and provide opportunities for infections caused by specific microorganisms, for example, by promoting the survival of Gram-negative intracellular bacteria.34
Biomechanical factors
Biomechanical factors, such as entheseal microtrauma called deep Koebner phenomenon, might also be involved in the pathogenesis of SpA,35 and resident immune cells might provide a link between a certain genetic predisposition, local tissue damage and the development of chronic inflammation.36 This hypothesis is supported by findings in certain mouse models, where strain release of the Achilles tendons resulted in decreased inflammation.37 Inflammation of the tendons occurs as a normal phenomenon after intense exercise (e.g. marathon running),38 while the progression to chronic inflammation remains unclear. Furthermore, a recently published study of patients with axSpA has shown exercise to be highly beneficial regarding disease activity as determined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI),39 but long-term effects on disease progression are evidently complicated by the often relatively slow nature of the disease.
Innate immunity
Despite broad, multifaceted evidence for associations with the genetic, environmental and biochemical factors described above, the aetiology of SpA remains unclear. The current evidence points to the involvement of common immunological pathways being involved in SpA as immune-mediated inflammatory diseases. This notion is partly based on the recognition that SpA does not share prototypical genetic, clinical and immunological features of prototypical T cell- and/or B cell-mediated autoimmune diseases such as RA and systemic lupus erythematosus.40 Furthermore, previous studies have identified polymorphisms in genes involved in innate immune recognition (CARD9) and cytokine signalling pathways, such as the TNF (TNF receptor superfamily member 1A, TNF receptor type 1-associated death domain protein, TNF receptor superfamily member 15), IL-1 (IL1A, IL1R2) and IL-23/IL-17 (IL-23R, signal transducer and activator of transcription 3) axis.41,42 SpA is, therefore, often considered an autoinflammatory disease caused by autoreactive innate immune cells rather than an autoimmune disease caused by autoreactive adaptive immunity.43 However, the autoinflammatory and autoimmune diseases are not distinct entities but may represent different ends of a spectrum from autoimmunity to autoinflammatory, with varying degrees of interactions or cross-talks by different mediators such as the complement system.36,44
Animal models
Various mouse and rat models of SpA are already well established but have individual shortcomings and might not fully mimic the disease in humans. However, these models provide the opportunity to investigate isolated and/or localized factors and pathophysiology within the disease. One of the first animal models introduced in axSpA was one with transgenic rats expressing HLA-B27 and human b2-microglobulin, which spontaneously resulted in a phenotype resembling human SpA-manifestations (rat-SpA), including effects within the gastrointestinal tract, peripheral and vertebral joints, male genital tract, skin, nails and heart.45 The severity of disease manifestations in the model is strongly dependent on a high number of both HLA-B27 and human b2-microglobulin transgene copies. Further studies showed a critical implication of myeloid-derived cells; the induction of rat-SpA was possible in irradiated wild-type rats by the transfer of hematopoietic stem cells originating from the bone marrow of a disease-prone HLA-B27 transgenic donor rat, and reciprocally HLA-B27 transgenic rats with established rat-SpA were cured by irradiation followed by transplantations of bone marrow cells from wild-type rats.46 A resembling observation has been described in a male patient diagnosed with r-axSpA and concomitant acute myeloid lymphoma.47 The patient experienced spontaneous regression of syndesmophytes after an allogeneic peripheral blood stem cell transplantation following a pre-transplantation conditioning regimen of total body irradiation and cyclophosphamide. The development of rat-SpA is highly dependent on the presence of CD4+ T cells but not CD8+ T cells.48 This observation challenges the arthritogenic peptide hypothesis regarding the role of HLA-B27 in the pathogenesis of SpA in humans.
Another important aspect of the transgenic rat model is the prerequisite interactions with microbes in the external environment. HLA-B27 transgenic rats raised under germ-free conditions do not develop gut and joint inflammation but can be induced by reconstitution with commensal bacteria.22 However, while IBD remains over-represented among patients with SpA, gut inflammation is not always present, and caution must be taken when extrapolating these findings to human conditions. Throughout the years, HLA-B27 has been investigated in various mouse models without success, and the focus has shifted after the introduction of efficient treatment options in human SpA. A mouse model with deletion of adenylate uridylate-rich elements in the murine TNF locus (so-called TNFΔARE mice) leads to spontaneous overexpression of mouse TNF and a phenotype resembling SpA,49 including polyarthritis and sacroiliitis, enthesitis, and ileitis resembling Crohn’s disease. Furthermore, differences in the presence of different types of TNF (soluble versus transmembrane [tmTNF]) have been studied in another mouse model (TgA86), which overexpress tmTNF due to a defect in A disintegrin and metalloprotease 17 (ADAM17). Under normal circumstances, ADAM17 cleaves tmTNF and thereby generates soluble TNF.50 A proteoglycan-induced mouse model in BALB/c mice phenotypically resembling r-axSpA with associated peripheral joint involvement was also developed almost three decades ago.51,52 This model was used recently to investigate the complement system in SpA. Inhibition of complement activation by targeting complement factor C3 was shown to be beneficial in terms of limiting structural damage associated with the disease by decreasing osteoblast activity within the spine.47 Furthermore, the deposition of complement factor C3b/iC3b and terminal complement complexes (TCC) (membrane attack complex; C5b-9) were elevated in the spinal bone marrow and intervertebral discs in mice with r-axSpA and decreased if complement activation was inhibited by targeting C3.
Most recently, a new model of axSpA, which involves the induction of collagen-induced arthritis and complement activation in C567BL/6 wild-type mice, has been published.53 This model involves the induction of severe arthritis by injecting five monoclonal anti-collagen antibodies followed by ilipopolysaccharide from Escherichia coli. The resulting phenotype had a large phenotypical resemblance with axSpA and involved the development of kyphosis, enthesitis and peripheral arthritis. Furthermore, histologic examinations have demonstrated chondrocyte proliferation, macrophage infiltration and complement factor C3 deposition at the kyphotic area, whereas no C3 deposition was observed in control animals.54 Animal models of complement involvement in axSpA provide exciting prospects for further investigation of both disease development and radiographic changes in axSpA, as complement has been shown to be involved in the bone remodelling processes.55
The complement system
The complement system consists of more than 40 soluble and membrane-bound proteins that constitute an essential part of the innate immune system.44 The complement proteins are germline-encoded and do not adapt to potential microbial challenges, in contrast to adaptive immunity. The complement system is based on an ancient immunological concept of pattern recognition: pattern recognition molecules (PRMs) recognize foreign structures (e.g. bacterial surfaces) or altered self-structures, which are called pathogen-associated molecular patterns and danger-associated molecular patterns, respectively.
Complement activation may proceed through three distinct pathways: the classical pathway (CP), the lectin pathway (LP) and the alternative pathway (AP).56 All three pathways converge in a shared terminal pathway and may result in the direct killing of the pathogen or in opsonization for elimination by phagocytosis and release of anaphylatoxins, i.e. C3a and C5a (Figure 2).56
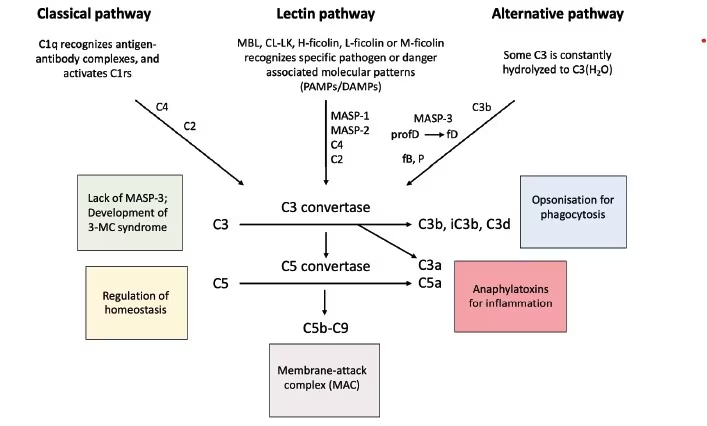
The classical pathway
The CP is primarily activated when the PRM complement factor C1q recognizes and binds Immunoglobulin M- or G-immune-complexes, such as antibodies bound to the surface of a pathogen. The binding process brings C1qrs complexes close to each other, which subsequently leads to the activation of the two proteases C1r and C1s. The activated C1s then cleaves C4 and C2, thereby generating the C3-convertase, C4bC2a, and further downstream complement activation.56,57
The lectin pathway
There are five PRMs in the LP: mannan-binding lectin (MBL), collectin-LK (a heteromer of CL-L1 [also called collectin-10] and CL-K1 [also called collectin-11]), H-ficolin, L-ficolin and M-ficolin (also called ficolin-3, ficolin-2 and ficolin-1, respectively). Each of the PRMs differs in various aspects, including ligand specificities, oligomeric structures and expression patterns. The PRMs circulate in plasma in complexes with three different serine proteases and two non-enzymatic proteins. For historical reasons, the proteases are named MBL-associated serine protease 1 (MASP-1), MASP-2 and MASP-3, and the two non-enzymatic MBL-associated proteins (MAps) are named MAp19 (also called sMAP) and MAp44 (also called MAP-1).58 The associated proteases and proteins of the LP are encoded by two genes: MASP-1, MASP-3 and MAp44 are encoded by the MASP1 gene, whereas MASP-2 and MAp19 are encoded by the MASP2 gene.59
The LP is activated when one of the PRMs binds to their respective ligand – danger-associated molecular patterns and pathogen-associated molecular patterns – on the surface of foreign pathogens and altered self-structures. This process results in the autoactivation of the adherent MASP-1, which subsequently activates MASP-2 in the neighbouring complexes of PRM/MASP-2.60 Activated MASP-2 cleaves complement factors C4 and C2, generating the C3 convertase, C4bC2a, and, as for CP, further downstream complement activation through the catalytic complement cascade in the common terminal pathway.61
The C3 convertases of the CP and LP hereby lead to (1) inflammation, mediated through the release of potent anaphylatoxins (i.e. C3a and C5a) and, consequently, the recruitment and activation of immune cells; (2) the deposition of complement fragments (i.e. C3b) and opsonization for phagocytosis; and (3) direct killing of the pathogen through the assembling of the plasma membrane penetrating and cell-killing membrane attack complex (MAC, also termed C5b-C9 or TCC). Therefore, all three effector mechanisms aim to eliminate the specific cause of complement activation.
The most recently discovered LP protease, MASP-3, is able to cleave pro-factor D to factor D, which is an essential part of the AP of the complement system.62 MASP-3 thereby provides a direct cross-link between the LP and AP. The non-enzymatic proteins MAp19 and MAp44 are able to form complexes with the PRMs and are thus thought to attenuate complement activation by competing with MASPs for binding to the PRMs.63
The alternative pathway
In contrast to the CP and LP, the AP is not based on a specific PRM-ligand interaction but is instead characterized by continuous activation. The C3 convertases of the AP are generated by the spontaneous hydrolysis of C3, resulting in the C3 convertase C3(H2O)Bb, or by C3b (which can originate from either LP or CP activation), which binds complement factor B, resulting in the C3 convertase C3bBb.56 MASP-3 cleaves pro-factor D to factor D, which is then able to cleave factor B, and properdin stabilizes the C3 convertase of the AP. AP activation thus functions as an amplification loop of complement activation. The AP activation is under normal conditions tightly controlled by several humoral and surface-related inhibitory mechanisms (i.e. complement factor I, complement factor H, CD46 and CD55) to prevent potential host-related harmful effects.56
Complement in spondyloarthritis
The interpretation and generalizability of the first studies of complement in SpA are challenged by only investigating CP and AP and focusing on single components of the complement system, as well as by subsequent historical changes in the differentiation of disease entities within the disease spectrum of SpA. Most studies only encompass patients with SpA as a unit and do not consider disease localization and/or extra musculoskeletal manifestations. An overview of the current studies is shown in Table 164–73 and further discusses in details in the following section.
Early studies of complement in SpA have shown divergent results regarding plasma levels of complement factors C1q, C3, C4, and C7 and total haemolytic complement concentration (i.e. CH50) compared with other inflammatory conditions, osteoarthritis and healthy individuals.64,65 A small study of nine patients with r-axSpA uncovered elevated levels of C9 compared with normal levels in healthy individuals.66 Complement activation, as examined by the levels of complement activation product C3d, was found to be elevated in patients with SpA compared with healthy controls.67 A study of 51 patients with SpA included in the PSoriatic arthritis, Ankylosing spondylitis, Rheumatoid Arthritis (PSARA) cohort (both r-axSpA and PsA) starting treatment with (1) TNF-inhibitor in monotherapy (n=25), (2) TNF-inhibitor in combination with methotrexate (n=10), or (3) methotrexate monotherapy (n=16) found baseline levels of soluble terminal complement complex (sC5–C9, also termed sTCC) to be at (for PsA) or above (for r-axSpA) normal reference levels.67 Levels of sTCC decreased significantly after the initiation of the TNF inhibitor ± methotrexate, whereas no significant changes were observed in patients receiving methotrexate monotherapy. The levels of sTCC remained stable between week 6 and week 26.
As described previously, the LP is the most recently described pathway of complement activation; hence, studies of LP in SpA are sparse and often limited by a focus on single components of complement activation. In a cross-sectional cohort of 89 Brazilian patients with SpA (including r-axSpA, PsA, reactive arthritis and undifferentiated arthritis), MBL levels were significantly lower compared with healthy individuals.69 The prevalence of MBL deficiency (≤100 ng/mL) was significantly higher in patients with SpA compared with healthy individuals (odds ratio [OR] 2.5; p=0.01). A new perspective on complement involvement in axSpA should focus on potential differences in r-axSpA and nr-axSpA, as the understanding of differences and similarities are currently limited: this might provide important insight into the pathogenesis of the two stages or phenotypes of the disease. Other perspectives should focus on complement involvement in specific SpA phenotypes and co-existing extra-musculoskeletal manifestations (e.g. acute anterior uveitis, IBD or psoriasis) to further elaborate the understanding of the pathogeneses.
Complement in axial spondyloarthritis
Recent studies of complement in axSpA emphasize the introduction of new treatment options and the clinical need for objective biomarkers for assessing disease activity, treatment response and prognosis (i.e. radiographic progression of the disease). Serum levels of complement factor C3 and C4 did not differ in 27 patients with biological DMARD r-axSpA divided according to disease activity (BASDAI <4 versus BASDAI ≥4) or clinical phenotype (with or without peripheral joint involvement) (Table 1).70 In 98 of the patients included in the clinical, multicentre randomized controlled trial GO-RAISE investigating treatment with golimumab in r-axSpA, baseline serum levels of complement factor C3 correlated with baseline Ankylosing Spondylitis Disease Activity Score with C-Reactive Protein (Spearman rho 0.53; p=0.000).71 Furthermore, reductions in complement factor C3 levels from baseline to week 14 correlated with an improvement in BASDAI score (Spearman rho 0.49; p=0.04). Baseline complement factor C3 levels were also associated with the development of fatty lesions assessed on magnetic resonance imaging at week 14 and week 104 (OR 0.13, p=0.001, and OR 0.15, p=0.008, respectively).
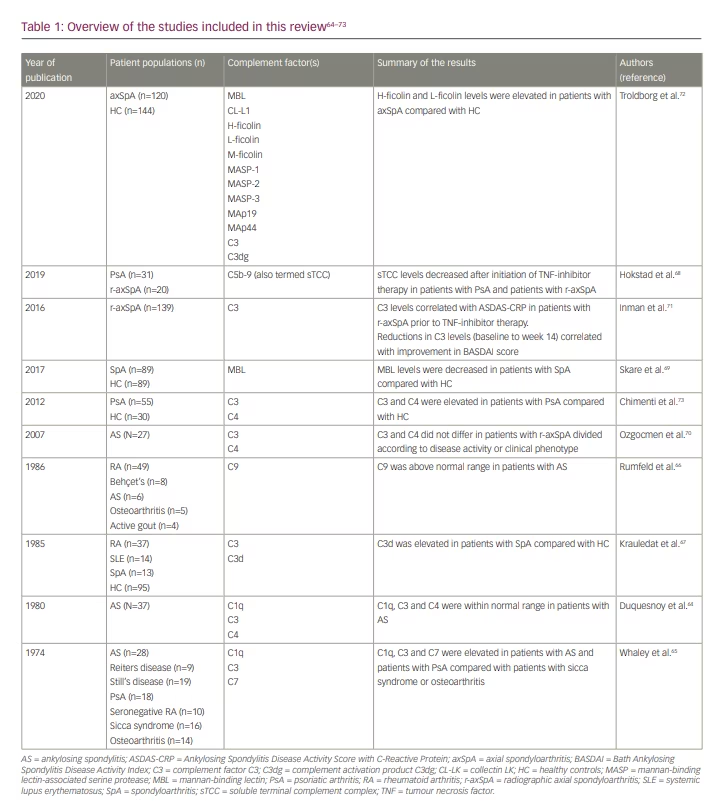
Another important aspect of research into axSpA regards its pathogenesis, which remains largely unexplained. A recently published cross-sectional study found complement LP proteins L-ficolin and H-ficolin to be elevated in patients with axSpA compared with age- and gender-matched healthy blood donors.72 As both of the PRMs, L-ficolin and H-ficolin, are well-established activators of the complement system through the LP, this provides new potential insights into the pathogenesis of axSpA.
Complement in spondyloarthritis
Chimenti et al. found serum levels of complement factors C3 and C4 to be elevated in 55 patients with PsA prior to the initiation of the TNF inhibitor compared with 30 healthy controls (Table 1).73 All patients with PsA fulfilled the ClASsification for Psoriatic ARthritis (CASPAR) criteria, had an inadequate clinical response to conventional synthetic DMARDs, and were randomized 1:1 to either adalimumab or etanercept. Twenty per cent of the patients received TNF-inhibitor monotherapy, whereas the remaining patients continued a combination therapy with methotrexate, sulfasalazine, ciclosporin or oral prednisolone 10 mg daily. Complement factor C3 and C4 serum levels decreased significantly after 22 weeks of treatment, irrespective of co-medications. Furthermore, patients with PsA with no or only mild European League Against Rheumatism (EULAR) response74 at week 22 had a significantly higher baseline levels of complement factor C3, suggesting its potential as a prognostic biomarker for the prediction of treatment response. Such interpretations are, however, limited by the use of the criteria for remission of RA in patients with PsA and not by the later developed Disease Activity in PSoriatic Arthritis (DAPSA) score nor by minimal disease activity (MDA).75,76
Conclusions
This review summarized the current knowledge regarding the involvement of complement in SpA, with axSpA and PsA as the main focuses of current studies. Complement activation was shown to be involved in animal models regarding the pathogenesis of axSpA,53,77 LP proteins H-ficolin and L-ficolin were shown to be elevated in patients with axSpA compared with healthy controls,71 and complement factor C3 was shown to decrease during treatment with TNF-inhibitor in patients with axSpA71 and patients with PsA,73 whereas C3 and C4 did not differ significantly according to disease activity in patients with axSpA.70 However, the field is challenged by the limited understanding of the pathogenesis of the diseases and, thus, the heterogeneity of the study populations included in the published studies, as well as the rapidly growing understanding of the functions of the complement system as serving functions beyond classical immunological defence.
The vast majority of investigations of biomarkers in patient cohorts are limited by the lack of testing whether changes represent general alterations; e.g. increases or decreases of protein production, associated with the disease or associated with a certain medical treatment of the disease. This limitation also applies to the current studies of biomarkers in SpA. However, the current development and progress regarding proteome analyses could shed light on such issues in the various conditions, including SpA. The current evidence regarding the involvement of innate immunity in SpA, and the unique capabilities of the complement system, which provides a cross-link between innate immunity, clearing of infections, homeostatic regulation, and human development, emphasizes that further investigations of complement in SpA is highly relevant. These future projects should address the lack of understanding of SpA pathogenesis, which poses a major obstacle to a correct and early SpA diagnosis and sufficient treatment of our patients with SpA, as well as striving to provide these investigations in clinically relevant settings. Topics of investigation that remain to be addressed further (1) complement activation in the pathogenesis of well-defined disease entities of SpA, (2) specific complement proteins as diagnostic biomarkers of early axSpA, (3) complement proteins as biomarkers of disease activity, (4) complement proteins as biomarkers for treatment response and (5) complement proteins as liquid biomarkers illustrating the progression of structural changes.